Article Text

Abstract
In order to identify a gene(s) susceptible to idiopathic pulmonary fibrosis (IPF), we conducted a genome-wide association (GWA) study by genotyping 159 patients with IPF and 934 controls for 214 508 tag single-nucleotide polymorphisms (SNPs). We further evaluated selected SNPs in a replication sample set (83 cases and 535 controls) and found a significant association of an SNP in intron 2 of the TERT gene (rs2736100), which encodes a reverse transcriptase that is a component of a telomerase, with IPF; a combination of two data sets revealed a p value of 2.9×10−8 (GWA, 2.8×10−6; replication, 3.6×10−3). Considering previous reports indicating that rare mutations of TERT are found in patients with familial IPF, we suggest that the common genetic variation within TERT may contribute to the risk of sporadic IFP in the Japanese population.
Statistics from Altmetric.com
Idiopathic pulmonary fibrosis (IPF), which is characterised by massive fibrotic changes and thickening of the alveolar walls in the lung, is a progressive lethal disease observed in people over 50 years of age.1 Pulmonary function in patients with IPF is very restricted due to reduced lung volumes and subsequent reduced capacity of gas exchange. More than 5 million people worldwide have been diagnosed with IPF, and the overall prognosis is dismal, with a median survival of 3–5 years after diagnosis.2 According to the international multidisciplinary consensus classification, there are seven distinct histological groups of idiopathic interstitial pneumonias (IIPs) with similar clinical presentations. IPF is the most common among the IIPs (approximately 40%).3 Although involvement of both genetic and environmental factors has been implicated, the exact mechanism of the disease remains unknown. As a result, to date, lung transplantation provides the only therapeutic opportunity for prolonged survival. Therefore, an understanding of the precise aetiology of the disease is especially valuable.
METHODS
Using Illumina HumanHap300 BeadChips (San Diego, California, USA; see Supplementary figs 1 and 2), we tested 214 508 SNPs for association with IPF in 159 patients with the disease, and 934 control subjects in Japan (Panel A; Supplementary methods). The call rates of all subjects were more than 0.99. After genotyping all subjects, SNPs with overall call rates of <0.99 were eliminated from the further analysis. Finally, we individually inspected the clustering plots of SNPs with a p value of <1.0×10−5 and excluded SNPs that showed a poor clustering pattern. To validate genotyping results of the Illumina assay, we genotyped all 159 case samples for Panel A with SNPs of a minimum p value <1.0×10−5 in the initial WGA study by using a multiplex polymerase chain reaction (PCR)-based Invader assay4 or the TaqMan assay (Applied Biosystems, Foster City, CA, USA). For rs2736100, call rates were 1.0 for case and control groups in the WGA study. The p values for the Hardy–Weinberg test were 0.55 and 0.14 for cases and controls, respectively.
All participants in this study gave informed consent for the collection of blood samples and clinico-pathological information.
RESULTS
The GWA analysis of 214 508 SNPs identified several candidate SNPs that might be associated with the disease (Supplementary table 1). A comparison of the case and the control groups identified nine SNPs with p values of <1.0×10−5 including one with a p value of <1.0×10−7.
To verify the findings obtained by the initial GWA study, we further evaluated these nine SNPs in an independent IPF case–control series. From the samples registered in BioBank Japan, we obtained 83 with IPF and 535 from normal controls (Panel B; Supplementary methods), and genotyped them by multiplex PCR and the subsequent Invader assay or TaqMan assay. In the additional case–control analysis, there was evidence of an association between the genotype of a SNP rs2736100 in the TERT gene and the disease risk (table 1 and Supplementary table 1). The combined p value of the two studies was 2.9×10−8, which was statistically significant after adjustment for multiple testing, using a conservative Bonferroni correction for the 214 508 tests. The odds ratio of the combined data was 2.11 (95% confidence interval (CI), 1.61 to 2.78). In the association testing from the GWA study covering a 500 kb region upstream and downstream of the locus, none of 64 SNPs tested revealed a significant association (data not shown). The linkage disequilibrium (LD) block, including rs2736100, is located within the TERT gene (fig 1).

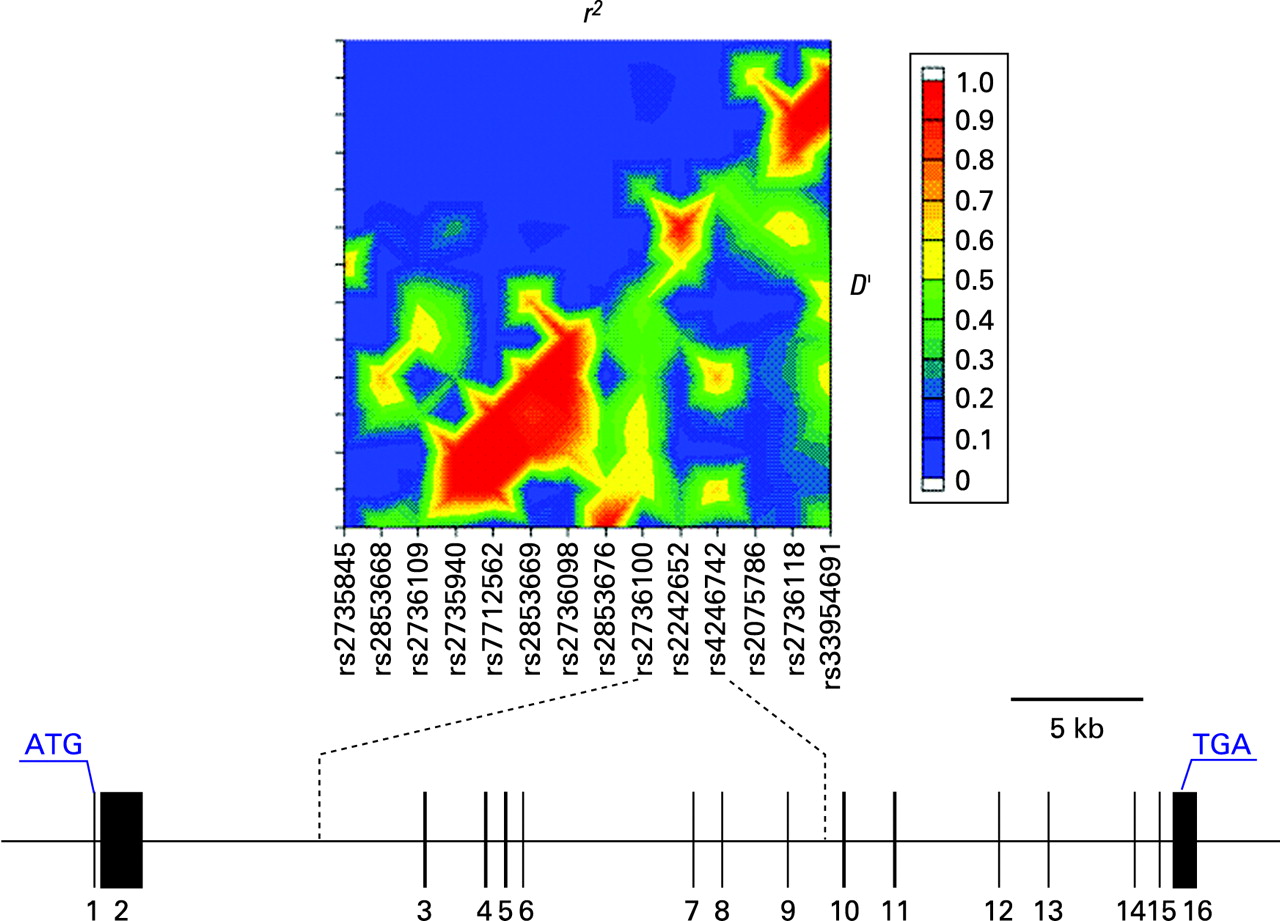{kind=link}
DISCUSSION
TERT encodes a reverse transcriptase that is a component of a telomerase.5 Telomerase is a specialised polymerase that adds telomere repeats (non-coding DNA (TTAGGG) elements) to the chromosomal ends. It has two essential components: a functional RNA (hTR), which contains a template region complementary to the telomeric sequence;6 and hTERT, which catalyses an addition of the telomeric repeats to the ends of the chromosomes.5 These components are important for the telomere replication and stabilisation against its shortening.7 While hTR is expressed in all tissues, hTERT is found to be highly expressed only in specific germ line cells, proliferative stem cells of renewal tissues, and immortal cancer cells.8–10 Mutations in these genes could cause shortening of the telomeres and impair the proliferative capacity of the telomerase-expressing cells; eg, mononuclear cells in the peripheral blood and other stem-like cells.
Several human diseases are known to be associated with dysfunction of telomerase. Heterozygous mutations of TERT or TERC, encoding hTR, have been found in patients with dyskeratosis congenita (DKC)11 12 and aplastic anaemia,13 suggesting the importance of telomere preservation and telomerase dosage for the maintenance of the tissue proliferative capacity relevant to the development of DKC and aplastic anaemia. Since the bronchoalveolar epithelium is a tissue of high turnover, similarly to the bone marrow and the skin, it relies on its local progenitor reserves that are limited by the telomere shortening.
Recently, two groups independently demonstrated that mutations in TERT were considered to be responsible for familial IPF in a Caucasian population14 15 on the basis that many affected people in the DKC families also had IPF.11 In all affected people, the telomere length in chromosomes derived from lymphocytes was less than 10% in comparison with age-matched controls.14 Tsakiri et al15 revealed missense and frame-shift mutations that co-segregated with IPF in the two families. They also identified five other mutations in TERT in additional families with IPF and demonstrated that all of the heterozygous carriers of TERT or TERC mutations had shorter telomeres than the age-matched family members without the mutations. Thus, collectively, these two studies indicate that the mutations in TERT decrease the stability of the telomerase complex and reduce the recruitment or activation of the telomerase, dramatically increasing the susceptibility to adult-onset IPF.
In this study, we demonstrated for the first time the potential role of a common genetic variation in TERT in sporadic IPF development. In fact, this study has two main limitations. First, there is an imbalance between the size of the case and control groups: the number of cases was nearly one-sixth of that of controls in both panels. Second, since the number of cases was small, our approach did not have sufficient statistical power to find all the genetic factors, but should be sufficient to identify a gene(s) having a relatively large effect like TERT. It is certain that some genes having small effects to the disease susceptibility were not detected. We suspect that the SNP identified (rs2736100) affects the expression level of TERT, or there are some other genetic variations linked to the same locus which probably alter the gene products and increase the risk of IPF. It is also possible that the risk of sporadic IPF associated with a common variation in TERT is under the influence of other genetic and/or environmental factors (such as smoking), some of which may be population-specific. Although the location of the, so far, unidentified variant in the TERT gene is still unknown, an extension of our investigation to other populations and to patients with familial IFP will be of value.
In conclusion, we suggest that the genetic variant in the TERT gene significantly contributes to susceptibility to sporadic IPF in the general population. Although further investigations will be required to validate the causative factor and clarify the biological mechanisms associated with IPF, our finding should shed on a light on the understanding of the IPF aetiology and the development of effective treatments.
Acknowledgments
We thank all the participants in the study, as well as all members of BioBank Japan, Institute of Medical Science, The University of Tokyo, and the SNP Research Center, The Institute of Physical and Chemical Research, for their contribution to the completion of this work.
REFERENCES
Supplementary materials
web only appendices 45/10/654
Files in this Data Supplement:
{kind=link}
{kind=link}
Footnotes
▸ Supplementary figs, tables and methods are published online only at http://jmg.bmj.com/content/vol45/issue10
Competing interests: None.
Ethics approval: Collection of blood samples and clinico-pathological information from patients and controls was undertaken with approval from the Ethical Committees at the SNP Research Center, The Institute of Physical and Chemical Research (RIKEN), Yokohama, Japan, and The Institute of Medical Science, The University of Tokyo, Tokyo, Japan.