Article Text

Abstract
Introduction: Autosomal dominant optic atrophy (ADOA) is considered as the most common form of hereditary optic neuropathy. Although genetic linkage studies point to the OPA1 locus on chromosome 3q28–q29 as by far the most common gene locus, previous screening studies—based on sequencing of the coding exons—detected OPA1 mutations in only 32–70% of ADOA patients. We therefore hypothesised that larger deletions or duplications that remained undetected in previous screening approaches may substantially contribute to the prevalence of OPA1 mutations in ADOA.
Methods: 42 independent ADOA patients were analysed for the presence of genomic rearrangements in OPA1 by means of multiplex ligation probe amplification (MLPA). Deletions or duplications were confirmed either by long distance polymerase chain reaction (PCR) and breakpoint sequencing or loss of heterozygosity analyses with flanking microsatellite markers. Patients underwent ophthalmological examination including visual acuity, colour vision testings, perimetry and funduscopy.
Results: We identified genomic rearrangements in 8 of 42 patients, including single exon deletions of exon 9 and exon 24, respectively, a deletion of exons 1–5, two different deletions of the complete OPA1 gene as well as a duplication of the exons 7–9, with the latter being present in three unrelated families. Patients’ phenotypes were highly variable, similar to patients with point mutation in OPA1.
Discussion: Our findings show that gross genomic aberrations at the OPA1 gene locus are frequent in ADOA and substantially contribute to the spectrum and prevalence of OPA1 mutations in ADOA patients. They further strengthen the hypothesis that haploinsufficiency is a major pathomechanism in OPA1 associated ADOA.
Statistics from Altmetric.com
Autosomal dominant optic atrophy (OMIM 165500) is the most frequent hereditary optic neuropathy, with an estimated prevalence of up to 1:12 000 in Denmark.1 2 It is characterised clinically by a variable loss of visual acuity, central or near central scotomas, reduced colour discrimination ability, and pallor of the optic nerve head.3 4 However, there is considerable intra- and interfamilial variability in progression and severity of the visual defects ranging from functionally asymptomatic carriers to legally blind patients.5–7 Histopathological examinations of affected donor eyes linked these visual impairments to a substantial loss of retinal ganglion cells and an atrophy of the optic nerve.8 9
Linkage analysis revealed the major gene locus for ADOA on chromosome 3q28–q2910 and subsequently we and others were able to identify a gene OPA1 (optic atrophy gene 1, RefSeq: NP_056375) that is mutated in ADOA families.11 12 Two further gene loci for ADOA, OPA4 on chromosome 18q12.2–q12.313 and OPA5 on chromosome 22q12.1–q13.1,14 have been mapped by linkage analyses, and dominant mutations in the OPA3 gene have been recently reported in families with ADOA associated with cataract,15 thus documenting genetic heterogeneity in ADOA.
The fractional prevalences of these multiple loci in ADOA still remain to be established, though it is commonly accepted that mutations in OPA1 are by far the most common cause of ADOA.
To date, 117 different pathogenic mutations in OPA1 have been described.16 Most of them are predicted to give rise to a truncated non-functional OPA1 protein. Therefore, haploinsufficiency is believed to be the major pathomechanism underlying OPA1 associated ADOA. This hypothesis is strengthened by the report of an ADOA family with a complete deletion of the OPA1 gene,17 and by recent experimental findings showing that OPA1 mRNA containing premature termination codons undergo nonsense mediated mRNA decay.18 Moreover, it has been shown in an OPA1 mouse mutant that mutant OPA1 protein is unstable in vivo.19
Among the few screening studies the reported prevalence of OPA1 mutations in unselected ADOA patients ranged between 32–70%.20–23 Since these studies relied on conventional single exon based screening strategies (that is, sequencing or indirect screening of polymerase chain reaction (PCR) amplified exons), gross chromosomal aberrations encompassing larger parts or the entire OPA1 gene could not be detected.
In this study we report for the first time on a screening for copy number variation and genomic rearrangements in the OPA1 gene by applying multiplex ligation probe amplification (MLPA) in a screen of 42 independent ADOA families, which were tested negative for OPA1 and OPA3 gene mutations by conventional qualitative screening techniques. Within this study group, we found eight cases carrying various forms of genomic rearrangements in the OPA1 gene or at the OPA1 gene locus that co-segregate with the disease in the respective families. Our findings indicate a high frequency of mutations involving genomic rearrangements of the OPA1 gene and that the prevalence of OPA1 mutations in ADOA is probably considerably higher than previously estimated.
PATIENTS AND METHODS
Patients
In this study we included 42 unrelated patients/families which were recruited at different clinical centres throughout Europe with a clinical diagnosis of dominant optic atrophy, based on ophthalmologic examinations and family history. Mutations in the OPA1 and the OPA3 gene could not be detected in these patients by means of either direct sequencing of the coding exons (with flanking intronic sequence) or DHPLC analysis. Clinical data of patients harbouring genomic rearrangements were compiled retrospectively or obtained upon reassessment, including visual acuity, colour vision performance, and findings in perimetry and funduscopy. The study was performed in accordance to the tenets of the Declaration of Helsinki and was approved by the local ethics committee.
Isolation of DNA
Genomic DNA was extracted from venous blood samples applying a standard salting-out procedure.
MLPA reaction and data extraction
MLPA reactions were performed using the P229 kit from MRC-Holland (Amsterdam, The Netherlands) following the manufacturers’ instructions and using an exactly defined amount of genomic DNA (100 ng). The obtained PCR products were separated on a 3100 Capillary Sequencer (Applied Biosystems, Darmstadt, Germany) and subsequently analysed using the Coffalyser spreadsheet macro (control probe analysis, MRC-Holland). Results were considered significant when relative peak areas for a probe were decreased or increased by at least 35%, respectively, in at least two independent experiments. The performance of the MLPA kit was evaluated with a DNA sample of a subject carrying an OPA1 gene deletion,17 kindly provided by Dr Carmel Toomes and Dr Chris Inglehearn, Leeds University, UK.
Assessment of genomic rearrangements
Genomic rearrangements were confirmed by long distance PCR reactions, all carried out with TaKaRa LA Taq reagents (Takara Bio Inc, Shiga, Japan) under recommended cycling PCR conditions and applying the following oligonucleotide pairs: Deletion exon 9: exon8-L: 5′-CCG TTT TAG TTT TTA CGA TGA AGA-3′; exon10-R: 5′-CTT TAA CTG TCT TTT ATA AGC TCA TCC-3′. Deletion exon 24: exon23-L: 5′-TTT TTC CTT TAT TTC AAC TGC C-3′; exon25-R: 5′-TTT CCC CAG ATG ATC AAA GG-3′. Duplication 7–9: Seq872L: 5′-GAT GTT CTC TCT GAT TAT GAT GCC-3′; Seq901R: 5′-CAG ATG ATC TTG CGT ATT ATA ACT GG-3′. PCR products were either directly sequenced after ExoSAP treatment (USB, Staufen, Germany) or separated on agarose gels and purified using QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). Sequencing was done using BigDyeTerminator Chemistry 1.1 (Applied Biosystems) and reactions separated on an ABI 3100 DNA Sequencer. For segregation analysis of intragenic rearrangements, breakpoint PCR reactions were established using the following oligonucleotide pairs: Del-Ex-9-L: 5′-AAA TTA GCC ATA TGC GTG AAA-3′ and Del-Ex-9-R: 5′-GAC AAA CCG GAA AGA GAC CA-3′; Del-Ex24-L: 5′-TCC ATT TAC AAG TTA ATT GTT TGC AT-3′ and Del-Ex24-R: 5′-CAT GTC TAG CAA CTC AAC ATT TCC-3′; Dupl-Ex7-9-L: 5′-TTC TTG ACA GGT TTG ATA TGG AGA-3′ and Dupl-Ex7-9-R: 5′-AAT TAA TGC ATG TCA GTG TCA CCT-3′, respectively. Breakpoint PCRs were performed using standard Taq polymerase (FIREPol, Solis Biodyne, Tartu, Estonia) and a short extension time of not more than 30 s.
Larger deletions encompassing the whole OPA1 gene were confirmed by genotyping of annotated microsatellite markers covering a region of 3.5 Mbp on chromosome 3q28–q29. The SNP rs281817 was amplified using the following primers: rs281817-L: 5′-GCA CAC AGG CTT TCC CTA AG-3′; rs281817-R: 5′-CAG AAC AGA TGC TGG GGA AT-3′ under standard conditions and subsequently sequenced. In addition we applied an established panel consisting of seven microsatellite markers located on different chromosomes to test for kindredship.
Exon and intron nomenclature
The designation of OPA1 exons used in this article is based on transcript variant 1 (Genbank entry NM_015560). To avoid equivocal interpretation we designated introns by specifying their location between exons (for example “intron 8–9”).
RESULTS
We selected 42 independent ADOA patients/families who had tested negative for mutations in OPA1 and OPA3 in conventional, qualitative screening procedures. Moreover, we only included patients with a positive family history of optic atrophy. For MLPA we used the current P229 kit that includes 23 probes for the OPA1 gene. These probes target 20 of the 31 OPA1 exons (including alternatively spliced exons). MLPA was carried out in triplicates for each tested patient. Copy number variations were considered significant when relative peak areas for a probe were decreased or increased by at least 35%, respectively, in at least two independent experiments. Applying this strategy we were able to detect copy number variations for single or multiple probes in eight independent index patients.
Heterozygous deletion of exon 9
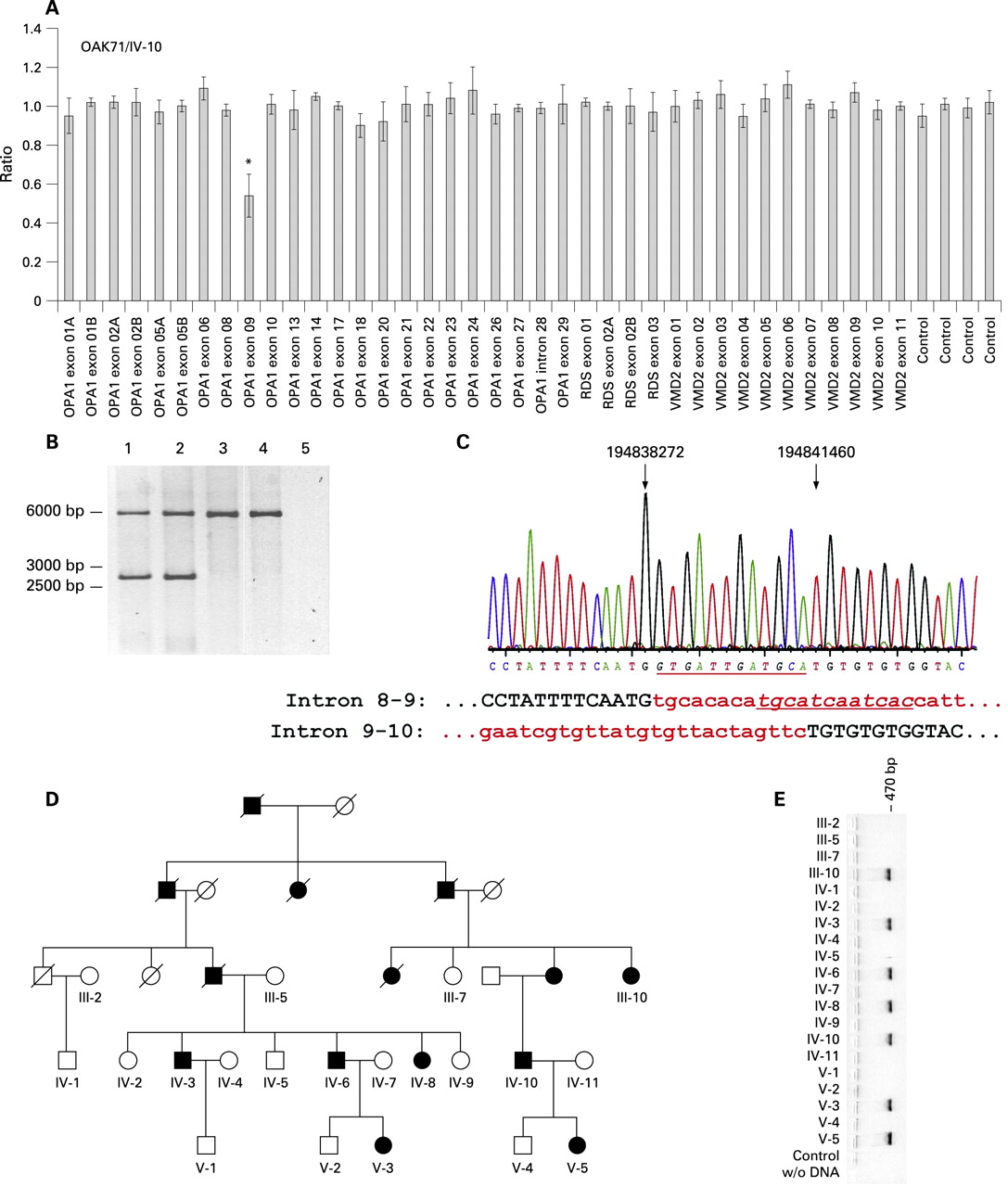
MLPA reaction revealed a significant decrease to about 50% for the exon 9 probe of the OPA1 gene in the index patient of a large ADOA family (OAK71, fig 1A). To confirm this deletion of exon 9 a long distance PCR was performed using primers located in exons 8 and 10 which give rise to an amplicon of 5888 bp from the wild type allele. Yet PCRs with DNA from the index patient (subject IV-10, fig 1B, lane 1) and another affected family member (subject IV-8, fig 1B, lane 2) revealed an additional smaller fragment of approximately 2.7 kb that was absent in an unaffected family member (subject IV-2, fig 1B, lane 3) and an unrelated healthy control (fig 1B, lane 4). Subsequent sequencing of the smaller band revealed deletion breakpoints in intron 8–9 (np 194,838,272, UCSC Human Genome Assembly of March 2006) and intron 9–10 (np 194,841,460, fig 1C). Therefore the deletion comprises a sequence of 3,188 bp of the OPA1 gene. Between the deletion breakpoints a dodecameric sequence (5′-gtgattgatgca-3′) is inserted that represents the reverse complement of a sequence eight nucleotides downstream of the breakpoint in intron 8–9 (fig 1C, underlined sequence). In silico analyses revealed a repetitive element in proximity to this sequence, while adjacent to the breakpoint in intron 9–10, there is a type I transposon element (not shown). Therefore it is likely that this dodecameric sequence represents a remnant of the deletion event.

The co-segregation of this mutation could be confirmed by a breakpoint specific PCR for all available patients from this family (fig 1D). The presence of a 500 bp amplicon indicates the exon 9 deletion being present solely in affected family members (fig 1E). This was confirmed by sequencing (not shown). The deletion of exon 9 results in an in-frame deletion of 38 amino acid residues (p.V291_K328del38) if we assume that splicing proceeds between exons 8 and 10. Exon 9 encodes part of OPA1’s GTPase domain, including the phosphate binding motif, and is a known hotspot for mutations in the OPA1 gene.16 18
Heterozygous deletion of exon 24
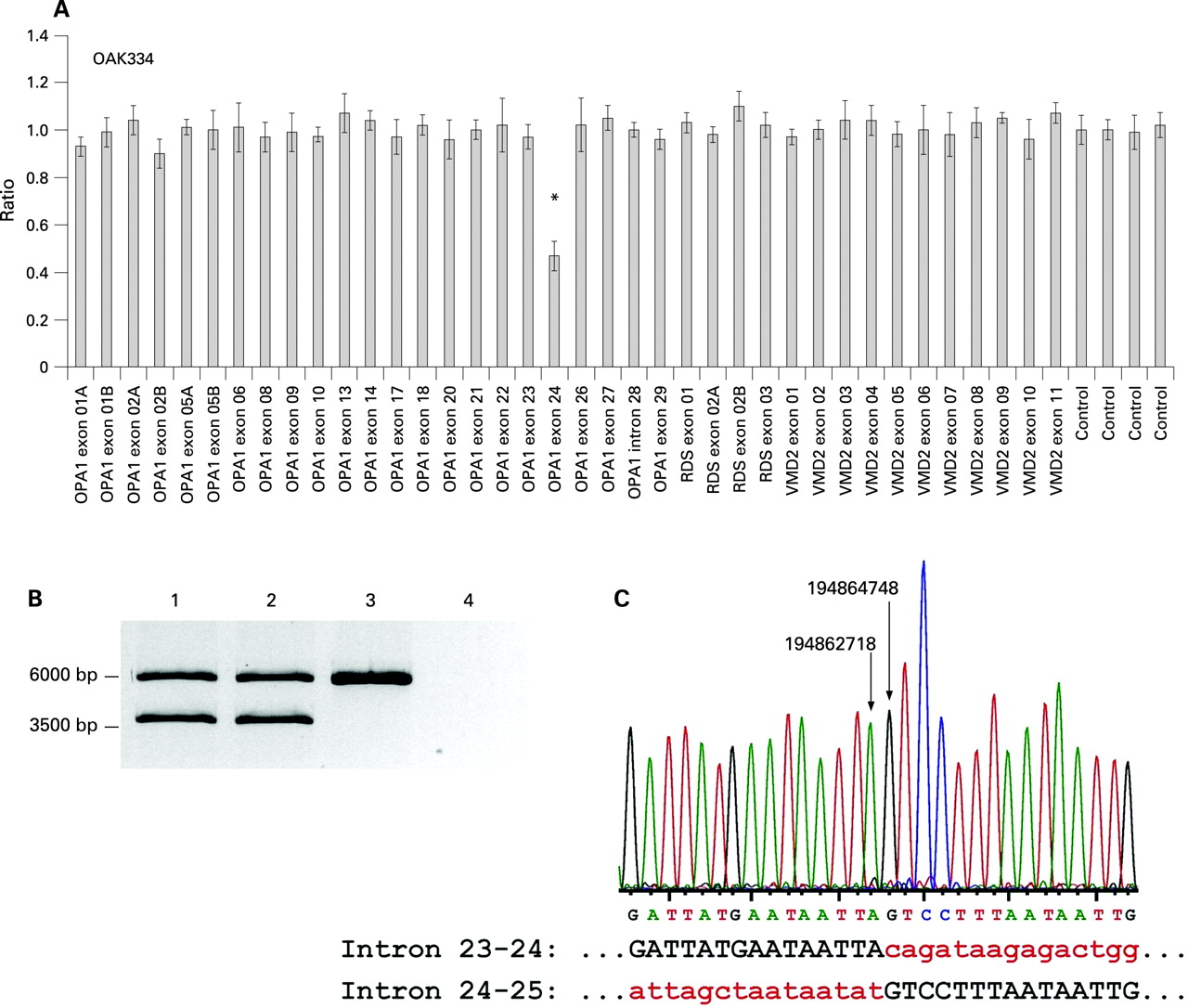
MLPA revealed a clear reduction to about 50% for the exon 24 probe in the index patient of the family OAK334 (fig 2A). Subsequent long range PCR with oligonucleotides located in exons 23 and 25 yielded a fragment of about 3.7 kb in addition to the expected 5.76 kb fragment for the wild type allele (fig 2B). This smaller fragment was also amplified with DNA from the affected uncle (fig 2B, lane 2), but was absent in controls. Sequencing of the 3.7 kb fragment via primer walking uncovered a deletion with breakpoints in intron 23–24 (np 194,862,718) and intron 24–25 (np 194,864,748; fig 2C). Therefore the deletion comprises 2030 bp of sequence including exon 24 and substantial proportions of the flanking introns. In silico analysis revealed type I transposon elements adjacent to both breakpoints (not shown). The deletion is predicted to result in an in-frame deletion of 47 amino acid residues (p.C786_L832del47) in the C terminal part of the dynamin central region of the OPA1 protein.

Heterozygous deletion of the exons 1–5
MLPA reaction uncovered a reduction of the exon 1–5 probes in the index patient of the family OAK506 to <60% (fig 3). The results were obtained three times for the index as well as for her affected father. No MLPA pattern change could be observed for the unaffected mother. In subsequent marker analyses we observed heterozygosity for markers D3S1601 to D3S3642, the latter defining the outermost boundary of the deletion (data not shown). Further telomeric markers including D3S3590 and four consecutive SNPs, rs281817, rs281811, rs189737, and rs169464, were not informative in the nuclear family. The deletion encompasses the first 208 codons of the open reading frame including the genuine start codon and the mitochondrial import sequence. It most likely also includes promoter sequences and hence affects transcription of the mutant allele.

Heterozygous deletions of the complete coding region of the OPA1 gene
The results of the MLPA reaction indicated a complete or near complete heterozygous deletion of the OPA1 gene in patients from two unrelated families (OAK230/II-1 and OAK103/II-1; fig 4A,B). In the patient OAK230/II-1 the two most 3′ probes targeting intron 28–29 and exon 29 were not reduced (fig 4A, OPA1 intron 28–29 and exon 29), indicating that a small proportion of the gene is still present in the mutated allele. In order to confirm this finding and to set the limit for centromeric extension of the deletion, we performed a microsatellite marker segregation analysis with three affected family members (fig 4C). Our results showed loss of heterozygosity (LOH) of the four successive markers D3S3669, D3S1523, D3S3642, and D3S3590. In addition, the father carries a different allele for these markers than his children. Since we confirmed parentship of the father applying a series of markers on other chromosomes (99% confidence interval, data not shown) we take this as proof for the deletion. Heterozygosity for the markers D3S1601 and D3S2305 defined the outermost borders of the deletion in this family, which encompasses at least 0.88 Mbp (between D3S3669 and OPA1 exon 27) and includes five or six further genes besides OPA1 (fig 4E).

For the patient OAK103/II-1, a complete deletion of the OPA1 gene can be concluded from the MLPA results (Figure 4B). The mother was referred as being asymptomatic, but is considered as gene carrier, since her sister (I-1) was diagnosed with ADOA and two other family members on the mother’s side (not available for this study) were reported to be affected as well. The deceased father of the index patient had no visual impairment nor is there any history of ocular disease in the father’s immediate family.
Upon segregation analysis we noted LOH for five successive microsatellite markers as well as for a single nucleotide polymorphism (SNP) in the ATP13A4 gene (fig 4D). Again, the presence of a different allele in the mother than in her daughter prove the deletion (parentship was confirmed with a 99% confidence interval, data not shown). The flanking markers D3S1601 and D3S1305 were heterozygous in at least two of the three analysed family members and thus define the outermost borders of the deletion. This indicates a deletion that ranges between 1.23 Mbp and 3.63 Mbp and encompasses the complete OPA1 gene and at least five additional genes (fig 4E).
Heterozygous duplication of exons 7 to 9 in three independent patients
Besides the deletions described above, we identified a duplication of exon 7 to 9 in three independent patients. In these patients the MLPA reaction revealed a significant increase of 140–150% of the probes for OPA1 exons 8 and 9 (fig 5A). Since the MLPA kit provides no probe set for OPA1 exon 7, the duplication of this exon could not be detected. Instead, we performed a long distance PCR using oligonucleotides placed both forward and reverse in exon 8 (fig 5C). Using this strategy, a specific PCR product of about 8 kb was amplified only in affected but not in unaffected individuals. Subsequent sequencing of this PCR product by primer walking uncovered the duplication insertion site at np 19,4840,568 in intron 9–10 (fig 5B). The duplication encompasses nominally 7,746 bp as its 5′ end maps to position np 194,832,822 in intron 6–7. Three additional nucleotides (gca) at the 5′ insertion site could be found three bases downstream of intron 9–10 and probably constitute a remnant of the insertion event (fig 5B, underlined sequence). Adjacent to both breakpoints in silico analysis revealed type I transposon elements. All three independent index patients showed the identical fusion sequence presented in fig 5B. On the protein level, this mutation would result in an in-frame duplication of 102 amino acid residues in the GTPase domain of the OPA1 protein (p.L227_K328dup102).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Applying a duplication specific PCR we were able to confirm co-segregation with the disease with an additional affected family member in family OAK46 and an asymptomatic carrier (fig 5D). In addition the same duplication was also present in family OAK66 and OAK144 as confirmed by sequencing (not shown).
Clinical phenotype of patients with genomic rearrangements in the OPA1 gene
Clinical data of patients from six families could be obtained and are compiled in table 1. All these patients showed typical features of ADOA, including reduced visual acuity, tritan defects, visual field defects and pallor of the optic disc, though highly variable in expression and severity. Even for the five patients with complete and near complete deletion of the OPA1 gene in which haploinsufficiency for OPA1 is obvious, we noted variable disease expression that was not correlated with age (for example, the mildly affected father I-1 in comparison with the severely affected son II:1 in OAK230). Moreover we observed an unaffected 52-year-old gene carrier in family OAK103 (fig 4). Additional ocular and extraocular affections were noted in some patients, but these do not segregate in the families and therefore most likely occur coincidentally.
DISCUSSION
Prevalences of OPA1 mutations in ADOA patients ranged between 32–70% in prior studies that did not take into account gross genomic rearrangements. This includes a study of our group in which OPA1 mutations were detected in 25 of 78 independent ADOA patients.21 In this paper we report the identification of eight subjects with genomic rearrangements at the OPA1 gene locus among a series of 42 patients that were tested negative for OPA1 mutations by conventional, qualitative screening analyses. Taking into account that this cohort was preselected, we calculate the prevalence of such genomic rearrangements to be 12.9% for all ADOA cases. This finding now considerably increases our total prevalence of OPA1 mutations to at least 45%. However, we reason that the fraction of genomic rearrangements is still an underestimate since the current MLPA kit only targets 20 of the 31 exons of the OPA1 gene. Moreover, other gross rearrangements that do not result in copy number changes like inversions still remain mostly undetected.
Using an exactly defined amount of genomic DNA and performing triplicate experiments, we found MLPA to be very reliable for the detection of copy number variations in OPA1. In fact, the positive results obtained were consistent in all three replications without any false positives and thus clearly exceeded our initial significance criterion. Furthermore, in all cases at least one additional affected family member was tested by MLPA and presented with the same result as the index patient.
There are currently a 117 different OPA1 mutations listed in the eOPA1 database (http://lbbma.univ-angers.fr/eOPA1/). Most of these mutations represent single case reports and only very few have been recurrently observed in more than a single family. In that context it is noteworthy that the same duplication of exons 7–9 identified here was present in three independent families. Although all three families are of German descent and therefore we cannot exclude a local founder effect, we believe that our results support the previous findings that the GTPase domain is a hotspot for mutations.16 18
The specific ocular pathology of OPA1 associated ADOA is not yet resolved. It has been argued that retinal ganglion cells (RGCs) may have an utmost high energy demand due to the unique feature that the initial portion of their axons is non-myelinated.24 A reduced amount of OPA1 which has been shown to participate in mitochondrial network homoeostasis and maintenance of the cristae structure may therefore primarily target RGCs.25 There are several lines of evidence which support this hypothesis of haploinsufficiency as a major pathomechanism in OPA1-associated ADOA. Most of the published mutations of the OPA1 gene are predicted to give rise to truncated polypeptides either by nonsense or frameshift mutations leading to premature termination codons (PTCs) or deletions and splicing mutations that result in in-frame deletions. Recent data from our group showed that PTC containing OPA1 transcripts are unstable, most likely due to nonsense mediated mRNA decay (NMD).18 Moreover investigations of a mouse mutant which expresses an in-frame deleted transcript demonstrated that such a shortened OPA1 protein is rapidly degraded in vivo.19 A similar effect might be attributed to the exon 9 and exon 24 deletions found in this study, which also presumably result in shortened polypeptides with in-frame deletions. Yet the proposed model of haploinsufficiency is most convincingly supported by large or complete gene deletions as found in three of our families. The mutant allele in family OAK506 is unlikely to be transcribed as the upstream deletion would adversely affect the mRNA transcription machinery. Furthermore, if transcribed it would still lack the mitochondrial import sequence.
The deletion in OAK230 encompasses almost the complete coding sequence with the exception that exon 28, which encodes the 21 C terminal amino acid residues, might still be present. In family OAK103 the OPA1 gene is completely deleted. A similar complete OPA1 gene deletion has been described by Marchbank and co-workers in an Australian ADOA family.17 All these three large deletions are different but encompass a common set of additional genes besides OPA1—namely ATP13A4, ATP13A5, HRASLS and the hypothetical gene LOC647319 (fig 3E). HRASLS is an HRAS-like suppressor gene that might play a role in the RAS signalling pathway26 and ATP13A4 and ATP13A5 encode putative cation transport (p-type) ATPases. Interestingly Kwasnicka-Crawford and co-workers recently found the ATP13A4 gene to be disrupted due to a paracentric chromosomal inversion in a girl with expressive and receptive language delay.27 However, neither such developmental delay nor any other co-segregating symptoms are documented in our families or reported for the Australian family, suggesting that haploidy for these genes does not cause a clinically manifesting phenotype. Moreover, there is no evidence that these chromosomal deletions aggravate the phenotypic expression of ADOA in these families. On the contrary, there are asymptomatic gene carriers in the Australian family as well as in our family OAK103. In this respect, it might be noteworthy that in two families with the duplication of exons 7–9 (OAK46 and OAK66), we know about at least one asymptomatic mutation carrier and a subject who did not become aware of his visual acuity problems until the age of about 40 years (OAK144). Similar to previous studies on patients with point mutations in OPA1, we noted a high variability of disease expression and severity among the affected subjects with genomic rearrangements. However, all patients described here showed the typical non-syndromic ADOA phenotype without any additional symptoms as described recently.28 29 This is in accordance with haploinsufficiency being the underlying pathomechanism in genomic rearrangements described herein, in contrast to gain-of-function-mutations being associated with an ADOA plus phenotype.28 29
In conclusion, our data demonstrate that genomic rearrangements of the OPA1 gene are a frequent cause of ADOA and contribute substantially to the prevalence of OPA1 mutations in this disorder. We would therefore like to recommend that analyses for structural rearrangements of the OPA1 gene, either by MLPA or other techniques, should be routinely performed in mutation screenings in ADOA subjects.
Acknowledgments
We are deeply grateful to all patients and family members and the referring clinicians for their contribution to this study. We also appreciate many helpful comments and discussions by Simone Schimpf and Tanja Grau. Finally, thanks to Dr Carmel Toomes and Dr Chris Inglehearn (Leeds University) for their reference DNA sample with an OPA1 deletion.
REFERENCES
Footnotes
Competing interests: None.