


Abstract
Background: In contrast to the several reports of alkaline extracts (Sasa-health, SE), no study of flavonoids from the leaves of S. senanensis has been reported. Four flavonoids were isolated from this plant species and their biological activities were investigated. Materials and methods: Luteolin 6-C-β-D-glucoside [1], luteolin 7-O-β-D-glucoside [2], luteolin 6-C-α-L-arabinoside [3] and tricin [4] were extracted from the leaf of S. senanensis with methanol, partitioned with ethyl acetate, separated by Sephadex LH-20 and purified by high-performance liquid chromatography (HPLC). The structure was determined by ultraviolet (UV) spectra, high-resolution mass spectra (HR-MS) and nuclear magnetic resonance (NMR). Results: The luteolin glycosides, 1-3 showed no cytotoxicity against the human normal oral cells and oral squamous cell carcinoma cell lines used up to 0.8 mg/ml, whereas 4 was highly cytotoxic. The luteolin glycosides 1-3 protected the cells from UV induced cytotoxicity, more efficiently than 4. The anti-HIV activity of 4 (Selectivity index, SI=27) was much higher than that of the luteolin glycosides (SI=2-7), but lower than that of SE (SI=40). The scavenging activity of 1-3 against 1,1-diphenyl-2-picrylhydrazyl (DPPH) and superoxide anion radicals was comparable with that of quercetin and, much higher than that of 4. Conclusion: The luteolin glycosides from S.senanensis show several new biological properties distinct from tricin and the anti-UV activity of the luteolin glycosides may be derived from their radical scavenging activity.
Plants in the Sasa genus are distributed only in the eastern Asia region. In China and Japan, the plants have been used as food and folk medicines, and the leaves have been used in Japan as tea or functional food. Systematic studies of the pharmacological effects of S. veitchii, have demonstrated antiseptic (1), membrane stabilising (2), anti-inflammatory and phagocytic (3), radical scavenging (4, 5), antibacterial and anti-viral (5). It has recently been reported that the alkaline extract of the leaves of Sasa senanensis Rehder (SE) inhibited the nitric oxide (NO) and prostaglandin E2 (PGE2) production by lipopolysaccharide (LPS)-activated mouse macrophage-like cells (RAW264.7) via inhibition of inducible NO synthase and cyclooxygenase-2 expression at both protein and mRNA levels (6). We have recently suggested that multiple components of SE may be associated with each other in the native state or after extraction with alkaline solution (7). Recently, the antioxidative activities of a mixture of arabinoxylan and lignins in S. senanensis have been reported (8). We have previously reported several lignin-like biological activities of the alkaline extract of the leaf of S. senanensis (Sasa-health) (5, 7). However, the flavonoids from the leaves of S. senanensis have not been studied yet. Therefore, flavonoid was isolated from S. senanensis and the biological activity was investigated.
Materials and Methods
Materials. The following chemicals and reagents were obtained from the indicated companies: Dulbecco's modified Eagle's medium (DMEM) (Gibco BRL, Grand Island, NY, USA); fetal bovine serum (FBS), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), hypoxanthine (HX), xanthine oxidase (XOD), diethylenetriaminepentaacetic acid (DETAPAC), 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), Roswell Park memorial institute 1640 (RPMI 1640) medium (Sigma Chemical Co. St. Louis, MO, USA); dimethyl sulfoxide (DMSO), sodium copper chlorophyllin power, dextran sulfate (5 kD) (Wako Pure Chemical Ind., Ltd., Osaka, Japan); curdlan sulfate (79 kD; Ajinomoto Co. Inc., Tokyo, Japan), quercetin (Kishida Chemical, Tokyo, Japan) and 1,1-diphenyl-2-picrylhydrazyl (DPPH) (Tokyo Kasei, Tokyo, Japan).
SE was prepared and supplied by Daiwa Biological Research Institute Co. Ltd., Kawasaki, Kanagawa, Japan. One ml of SE was freeze dried to give the powder (52.3 mg).
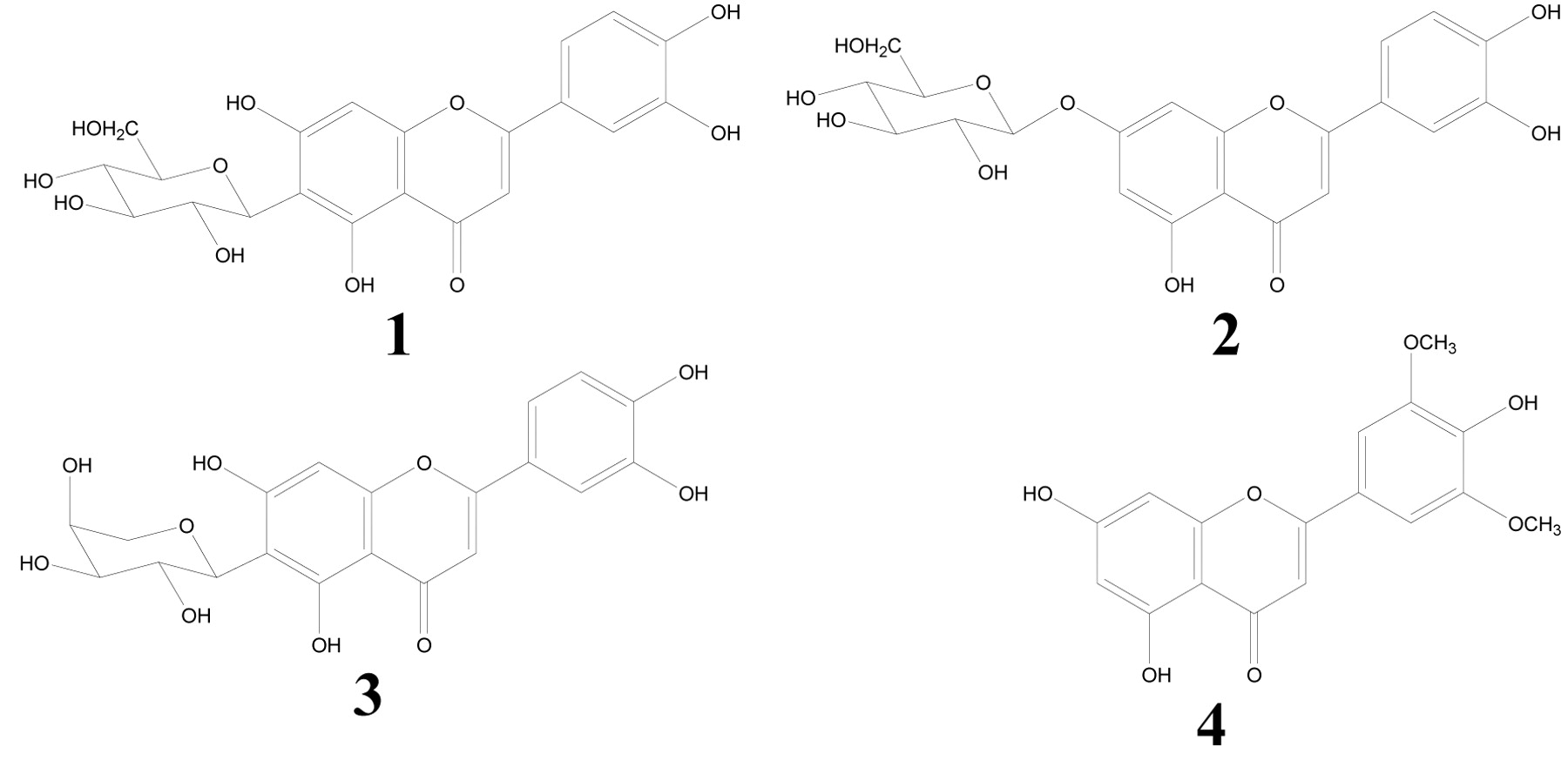
Isolation of luteolin glycosides and tricin. The protocol of the preparation of luteolin glycosides and tricin is outlined in Figure 1. The leaves of S. senanensis (200 g) were extracted with methanol (1 L). The methanol was removed by evaporation in vacuo at 40°C to obtain the methanol extract (14.44 g). The extract was suspended in water (400 ml) and partitioned with ethyl acetate and the ethyl acetate soluble portion was separated by Sephadex LH-20 (4.0 cm i.d. ×100 cm, GE Healthcare UK Ltd., Buckinghamshire, England) and eluted with 60% methanol to obtain eight fractions (Frs. 1-8). Frs 6 and 7 were further purified by preparative high-performance liquid chromatography (HPLC). The HPLC system used was composed of a Shimadzu LC-10AD pump and a Shimadzu SPD-M10AVP photodiode array detector (Shimadzu Co., Kyoto, Japan). The separation column was TSK gel ODS-80T (21.5 mm i.d. ×300 mm, Tosoh Co., Tokyo, Japan) with solvent H2O: acetonitrile: formic acid (70:20:10:1). Luteolin 6-C-β-D-glucoside [1] (14.0 mg) was obtained from Fr. 6. Luteolin 7-O-β-D-glucoside [2] (0.7 mg) and luteolin 6-C-α-L-arabinoside [3] (5.8 mg) were obtained from Fr. 7. HPLC showed that tricin [4] (9.2 mg) was the sole component of Fr. 8.
Structural analysis. Luteolin 6-C-β-D-glucoside [1]: yellow amorphous powder, [α]25D +30.7° (c=0.12, CH3OH), mp 232° (dec.), ultraviolet (UV) λmax (MeOH) nm (ε): 348 (22,200), 270 (17,600) and 258 (17,400). Electrospray ionization time of flight mass spectra (ESI-TOF-MS) m/z: 448 ([M+H]+), high-resolution mass spectra (HR-MS) m/z: 449.1094 (calcd. for C21H21O11, 449.1084).
Luteolin 7-O-β-D-glucoside [2]: yellow amorphous powder, [α]25D-81.1° (c=0.10, CH3OH), mp 261° (dec.), UV λmax (MeOH) nm (ε): 346 (20,500) and 270 (18,400). ESI-TOF-MS m/z: 448 ([M+H]+), 287 ([aglycon+H]+), HR-MS m/z: 449.0976 (calcd. for C21H21O11, 449.1084).
Luteolin 6-C-α-L-arabinoside [3]: yellow amorphous powder, [α]25D +66.0° (c=0.11, CH3OH), mp >300° (dec.), UV λmax (MeOH) nm (ε): 348 (22,100), 270 (17,600) and 258 (17,400). ESI-TOF-MS m/z: 419 ([M+H]+), HR-MS m/z: 419.1027 (calcd. for C20H19O10, 419.0978).
Tricin [4]: yellow amorphous powder, UV λmax (MeOH) nm (ε): 349 (41,000) and 269 (27,200). ESI-TOF-MS m/z: 331 ([M+H]+): HR-MS m/z: 331.0837 (Calcd. for C17H15O7, 331.0818) (Figure 2).
Cell culture. Human promyelocytic leukemia HL-60 cells were provided by Prof. K. Nakaya, Showa University, Japan. Human oral squamous cell carcinoma cell lines (HSC-2, HSC-3, HSC-4) were provided by Prof. M. Nagumo, Showa University. Normal human oral cells, gingival fibroblast (HGF), pulp cells (HPC) and periodontal ligament fibroblast (HPLF), were prepared from periodontal tissues, according to the guideline of the Intramural Ethic Committee (No. A0808), after obtaining the informed consent from a 12 year-old patient at the Meikai University Hospital. Since normal oral cells have a limited lifespan of 43-47 population doubling levels (PDL) (8), they were used at 8-15 PDL. The HL-60 cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin G and 100 μg/ml streptomycin sulfate under a humidified 5% CO2 atmosphere. The other cells were cultured in DMEM medium supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin G and 100 μg/ml streptomycin sulfate. The normal cells were detached with 0.25% trypsin-0.025% EDTA-2Na in phosphate-buffered saline without Mg2+ and Ca2+ (PBS(−)) and subcultured at a 1:4 split ratio once a week, with a medium change in between the subcultures. The tumor cell lines were similarly trypsinized and subcultured.

Purification of luteolin 6-C-β-D-glucoside [1], luteolin 7-O-β-D-glucoside [2], luteolin 6-C-α-L-arabinoside [3] and tricin [4].
Assay for cytotoxic activity. The cells (3×103 cells/well, 0.1 ml/well) were seeded in 96-microwell plates (Becton Dickinson, Franklin Lakes, NJ, USA) and incubated for 48 h to allow cell attachment. Near-confluent cells were treated for 48 h with various concentrations of the test compounds in fresh medium. The relative viable cell number of adherent cells (except for HL-60 cells) was then determined by the MTT method. In brief, control and sample-treated cells were incubated for 4 h with 0.2 mg/ml of MTT in the culture medium. After removing the medium, the reaction product, formazan, was extracted with DMSO and the absorbance (the relative viable cell number) was measured at 540 nm by a microplate reader (Multiskan Bichromatic Labsystems, Helsinki, Finland). The viability of the suspended cells, i.e. HL-60, was determined by cell counting with a hemocytometer after staining with 0.15% trypan blue. The 50% cytotoxic concentration (CC50) was determined from the dose-response curve.
Assay of anti-UV activity. HSC-2 cells, that showed the highest sensitivity against UV irradiation among 6 adherent cell lines tested (9, 10), were inoculated into 96-microwell plates (3×103 cells/well, 0.1 ml/well) and incubated for 48 h to allow cell attachment. The culture supernatant was replaced with 100 μl PBS(−) that contained various concentrations of the test substances, placed at 21 cm distance from a UV lamp (wavelength: 253.7 nm) and exposed to UV irradiation (6 J/m2/min) for 1 min. The cells were then incubated for a further 48 hours in DMEM containing 10% FBS to determine the relative viable cell number by the MTT method, as described above. From the dose-response curve, the 50% cytotoxic concentration (CC50) and the concentration that increased the viability of UV-irradiated cells to 50% of the untreated control level (EC50) were determined. The selectivity index (SI) was determined by the following equation: SI=CC50/EC50 (9, 10).
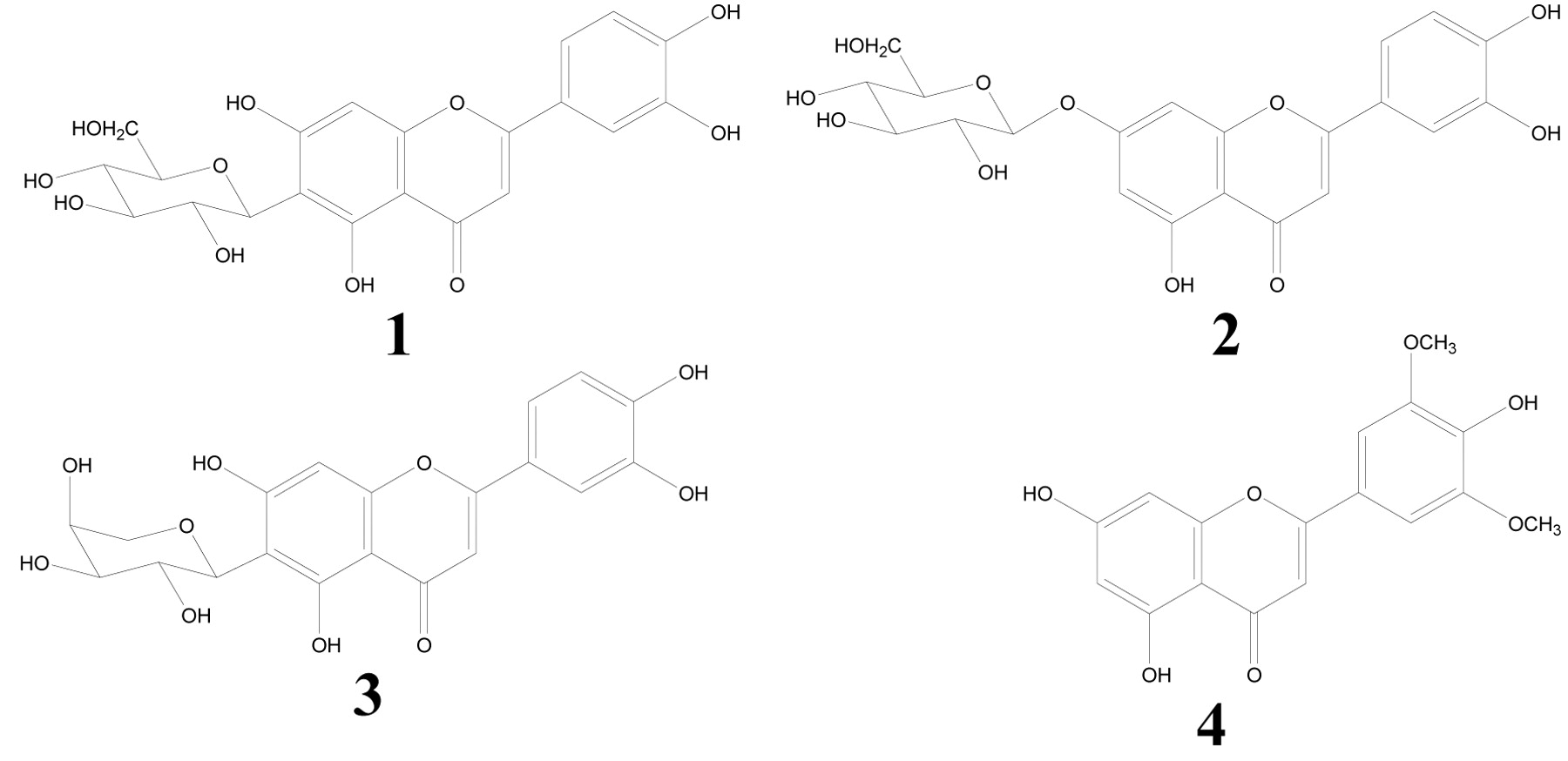
Structure of compounds. Luteolin 6-C-β-D-glucoside [1], luteolin 7-O-β-D-glucoside [2], luteolin 6-C-α-L-arabinoside [3] and tricin [4].
Assay for anti-HIV activity. Human T-cell leukemia virus I (HTLV-I)-bearing CD4 positive human T cell line, MT-4 cells, were cultured in RPMI 1640 medium supplemented with 10% FBS and infected with HIV-1IIIB at a multiplicity of infection of 0.01. The HIV- or mock-infected (control) MT-4 cells (3×104 cells/96-microwell) were incubated for five days with various concentrations of test samples and the relative viable cell number was determined by MTT assay (11). The CC50 and the EC50 were determined from the dose-response curve with mock-infected or HIV-infected cells, respectively. The anti-HIV activity was evaluated by the SI, which was calculated by the following equation: SI=CC50/EC50 (10).
Radical scavenging activity. The radical intensity was determined at 25°C, using electron spin resonance (ESR) spectroscopy (JEOL JES REIX, X-band, 100 kHz modulation frequency, JEOL Ltd., Tokyo, Japan) (12). The instrument settings were: centre field, 335.5±5.0 mT; microwave power, 16 mW; modulation amplitude, 0.1 mT: gain, 630; time constant, 0.03 sec and scanning time, 2 min.
The sample (100 μl) was added to 100 μl of 0.5 mM DPPH in ethanol. After the mixture was left to stand for 45 sec at room temperature, its ESR spectrum was measured.
For the determination of the superoxide anion (in the form of DMPO-OOH), produced by the HX-XOD reaction (total volume: 200 μl) (2 mM HX in 0.1 M phosphate buffer (PB) (pH 7.4) 50 μl, 0.5 mM DETAPAC 20 μl, 8% DMPO 30 μl, sample (in PB) 40 μl, PB 30 μl, XOD (0.5 U/ml in PB) 30 μl), the time constant was changed to 0.03 sec (12).
For the determination of the hydroxyl radical (in the form of DMPO-OH), produced by the Fenton reaction (200 μl) (1 mM FeSO4 (containing 0.2 mM DETAPAC) 50 μl, 0.1 M PB (pH 7.4) 50 μl, 92 mM DMPO 20 μl, sample (in H2O) 50 μl, 1 mM H2O2, 30 μl), the gain was changed to 160 (12).
The radical intensity of NO, produced from the reaction mixture of 20 μM carboxy-PTIO and 60 μM NOC-7, was determined in 0.1 M PB (pH 7.4) in the presence of 30% DMSO (microwave power and gain were changed to 8 mW and 400, respectively). When NOC-7 and carboxy-PTIO were mixed, NO was oxidised to NO2 and carboxy-PTIO was reduced to carboxy-PTI, which produces seven-line signals. The samples were added 3 min after mixing. The NO radical intensity was defined as the ratio of the signal intensity of the second peak to that of MnO (12).
Statistical analysis. Each value represents the mean±S.D. from triplicate assays.
Results
Cytotoxicity. Luteolin 6-C-β-D-glucoside, 1, luteolin 7-O-β-D-glucoside, 2 and luteolin 6-C-α-L-arabinoside, 3 showed no cytotoxicity against any of the oral normal cells (HGF, HPC, HPLF), the carcinoma cell lines (HSC-2, HSC-3, HSC-4) or the HL-60 cells, up to 800 μmg/ml. On the other hand, tricin, 4 was cytotoxic to the HSC-4 and HL-60 cells (Table I).
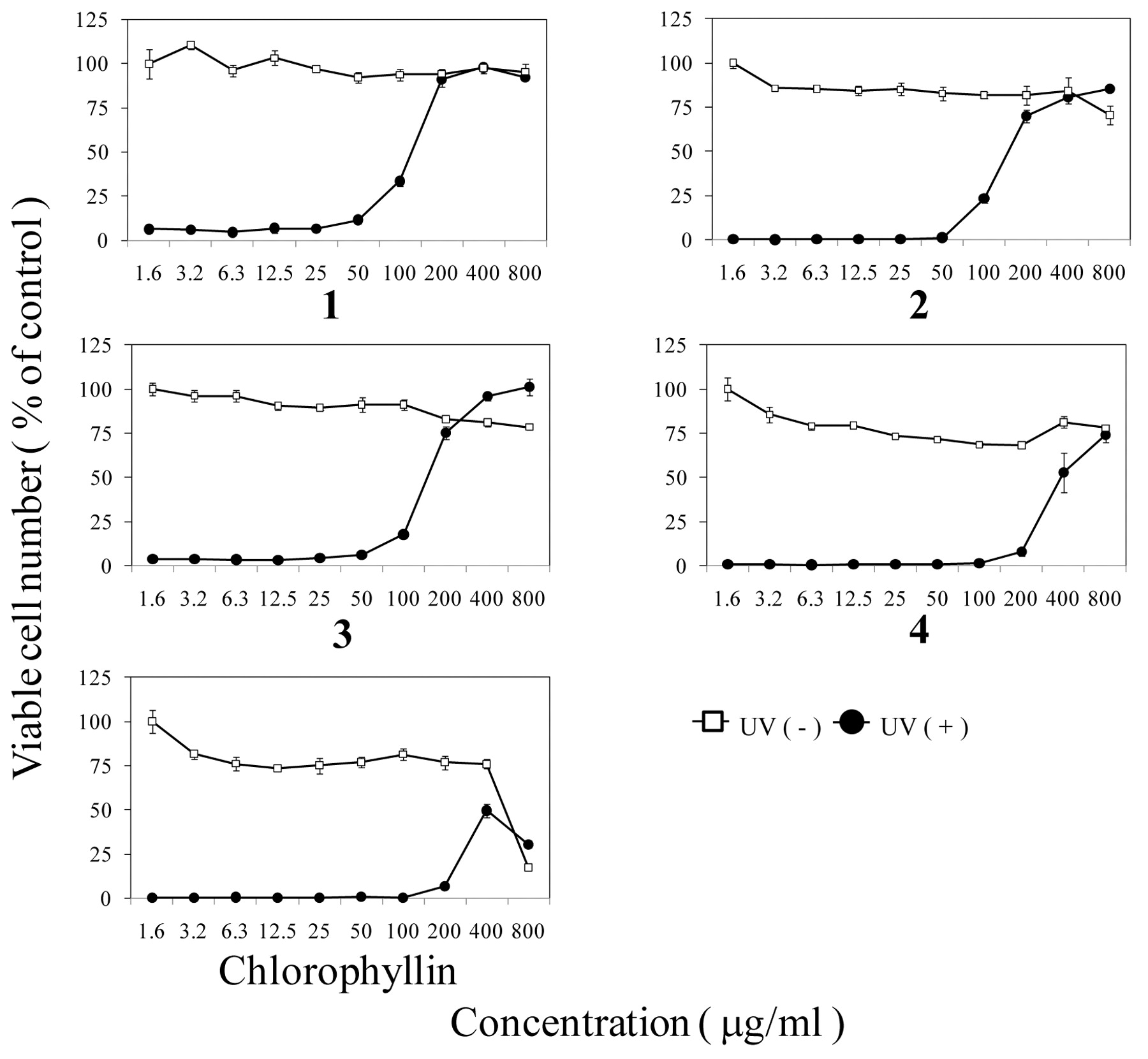
Anti-UV activity. The luteolin glycoside, 1-3, above 200 μg/ml, protected the HSC-2 cells from UV-induced cytotoxicity (Figure 3). The SI of these compounds was calculated to be >7.9, >6.0 and 6.2, respectively (Table II). The tricin, 4 showed anti-UV activity above 800 μg/ml, with a lower SI value (=>2.7). Chlorophyllin showed a much lower SI value (SI=0.53), due to its higher cytotoxicity. On the other hand, SE showed the highest SI value (SI=19.4) (Table II).
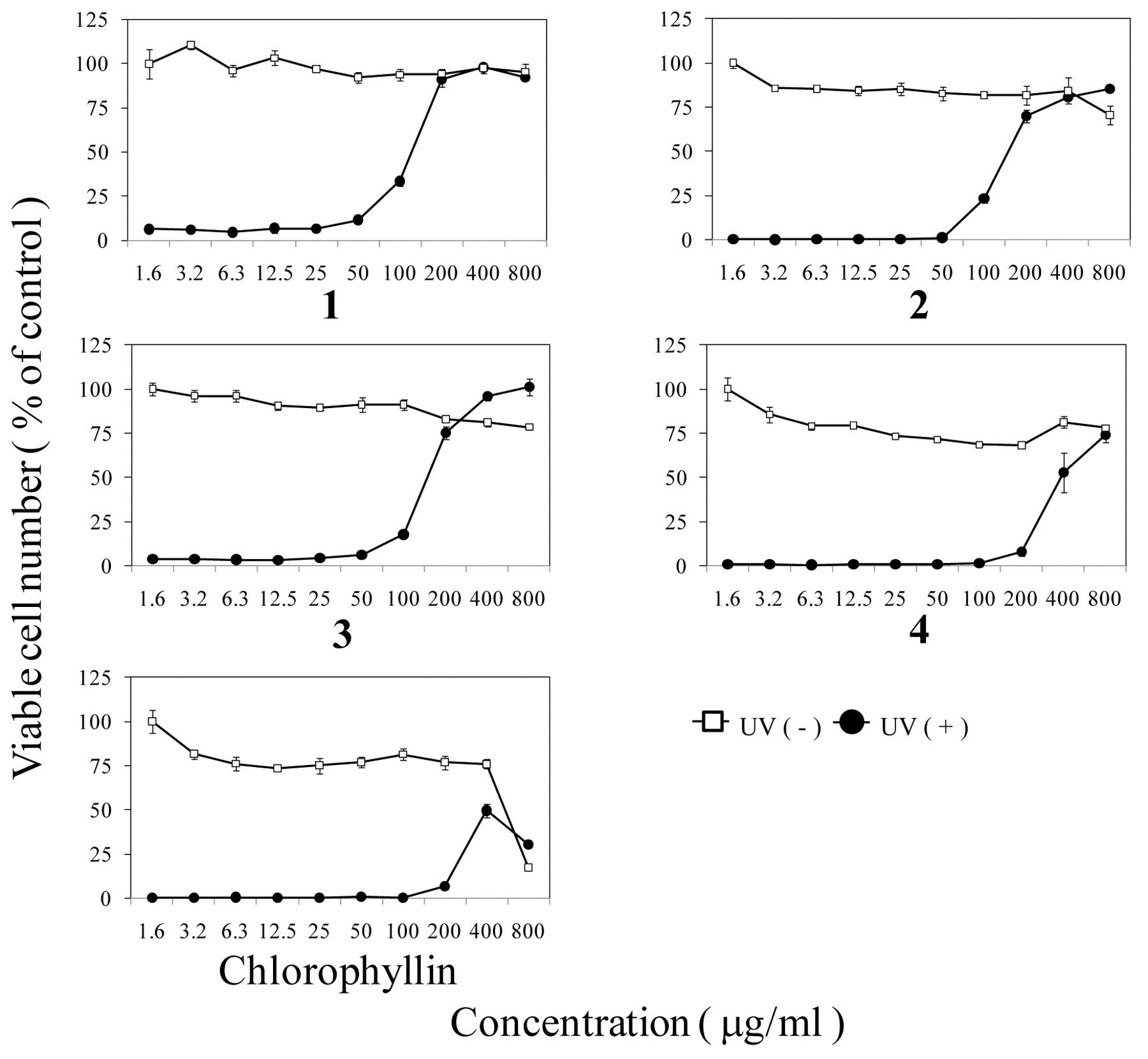
Anti-UV activity of compounds 1-4 and chlorophyllin. HSC-2 cells were exposed or not to UV irradiation in PBS(−) containing the indicated concentrations of each compound. Viable cell number was determined by MTT method 48 hours after irradiation, expressed as % of control non-irradiated and incubated without compound. Mean±S.D. of triplicate determinations.
Cytotoxic activity against human normal cell line and cancer cells line by luteolin glycosides and tricin.
Anti-HIV activity. The luteolin glycosides, 1-3, showed some anti-HIV activity (SI=>2, 7, >7), although their anti-HIV activity was much lower than that of the positive controls, such as dextran sulfate (DS) and curdlan sulfate (CRDS) (SI=137919, >133305) (Table III). The tricin, 4, showed somewhat higher anti-HIV activity (SI=24) than the luteolin glycoside, 1-3, but much lower than that of SE (SI=40) (Table III).
Radical scavenging activity. The luteolin glycosides, 1-3 showed about 20-30-fold higher DPPH radical scavenging activity than tricin, 4. The DPPH scavenging activity of 1 was slightly higher than that of quercetin (Table IV).
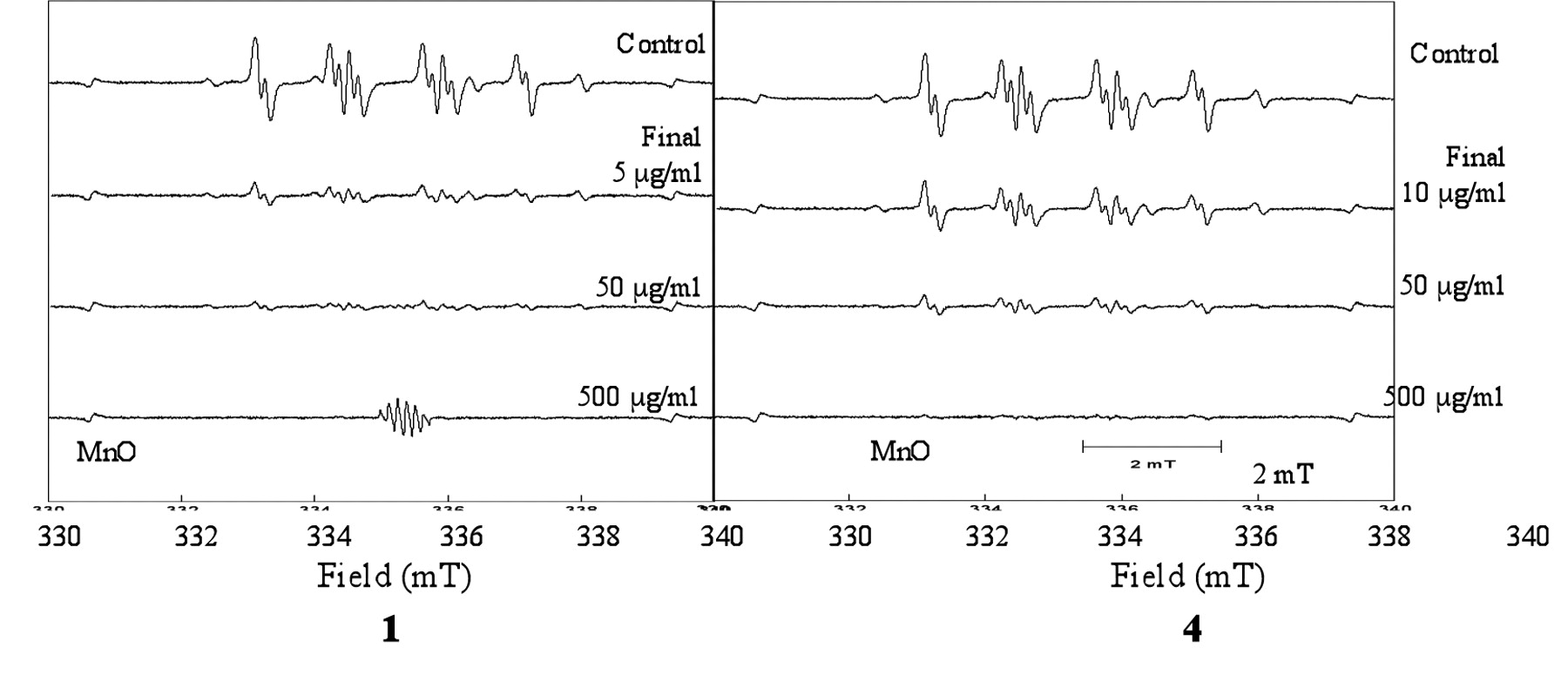
The luteolin glycoside, 1-3, showed about 5-10-fold higher superoxide anion radical scavenging activity than tricin, 4. Representative ESR spectroscopic patterns are shown in Figure 4. The superoxide anion scavenging activity of 1 was slightly higher than that of quercetin (Table IV).

Effect of 1 and 4 on the radical intensity of superoxide anion radical, measured as DMPO-OOH produced by hypoxanthine-xanthine oxidase reaction.
Anti-UV activity of luteolin glycosides and tricin.
Since the luteolin glycosides, 1-3, and tricin, 4, themselves scavenged the carboxy-PTIO, the NO scavenging activity could not be accurately determined (data not shown). DMSO, used to dissolve these compounds, interfered with the determination of hydroxyl radical scavenging activity (data not shown).
Discussion
The present study demonstrated several distinct biological properties of the luteolin glycosides, 1-3 and tricin, 4. First, the luteolin glycosides showed much lower cytotoxicity than tricin (Table I).
Tricin derivatives, such as tricin-7-O-β-(6’’-methoxy-cinnamic)-glucoside (13), tricin-4’-β-O-D-glucopyranoside, tricin-5-O-β-D-glucopyranoside (14), have been reported to show higher DPPH radical scavenging activity than Trolox or ascorbic acid. In the present study, the luteolin glycosides, 1-3, showed higher anti-UV activity (Table II) and radical scavenging activity (Table IV) than tricin, 4, suggesting a possible connection between these two activities. Furthermore, this is the first report that luteolin 6-C-α-L-arabinoside, 3, showed potent radical scavenging activity.
Anti-HIV activity of luteolin glycosides and tricin.
Radical scavenging activity of luteolin glycosides, tricin and quercetin (positive control).
Rice-Evans et al. (15) and Cao et al. (16) studied the relationship between the antioxidative activities and structure of flavonoids and concluded that the hydroxyl groups in the C-3’ and C-4’ of the B-ring are essential for the antioxidative activity, and that the double bond at C-2 and C-3 and the hydroxyl groups at C-3 enhance the antioxidative activity.
Tricin, 4, showed slightly higher anti-HIV activity (SI=24) than the luteolin glycosides, 1-3 (SI=2-7) (Table III), consistent with a previous report that tricin, 4, showed extremely high anti-human cytomegalovirus activity (17). However, the anti-HIV activity of tricin, 4 was lower than that of SE (Table III). This suggests that SE contains a lignin-carbohydrate complex that has high molecular mass (2nd and 3rd peaks in gel filtration chromatography (18) and water-solubility. Further purification of the anti-UV (18) and anti-HIV substances of SE and confirmation of their biological activity are underway.
- Received March 26, 2011.
- Revision received May 23, 2011.
- Accepted May 25, 2011.
- Copyright © 2011 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Effects of Kumaizasa (Sasa senanensis) Leaf Extract on Innate Immune Regulation in HEK293 Cells and Macrophages
- Antiviral and Antitumor Activity of Licorice Root Extracts
- Synergism of Alkaline Extract of the Leaves of Sasa senanensis Rehder and Antiviral Agents
- Prominent Anti-UV Activity and Possible Cosmetic Potential of Lignin-carbohydrate Complex
- Anti-Halitosis Effect of Toothpaste Supplemented with Alkaline Extract of the Leaves of Sasa senanensis Rehder
- Biological Interaction between Sasa senanensis Rehder Leaf Extract and Toothpaste Ingredients
- Anti-UV Activity of Lignin-Carbohydrate Complex and Related Compounds
- Structural Characterization of Anti-UV Components from Sasa senanensis Rehder Extract
- Pilot Clinical Study of Sasa senanensis Rehder Leaf Extract Treatment on Lichenoid Dysplasia
- Biological Activity of SE-10, Granulated Powder of Sasa senanensis Rehder Leaf Extract
- Comparative Study of Biological Activity of Three Commercial Products of Sasa senanensis Rehder Leaf Extract