


Abstract
IDIOPATHIC PULMONARY FIBROSIS (IPF) is a progressive and lethal pulmonary fibrotic lung disease. The diagnostic histological changes are called usual interstitial pneumonia and are characterized by histological temporal heterogeneity, whereby normal lung tissue is interspersed with interstitial fibrosis, honeycomb cysts and fibroblast foci. Pulmonary functions show restricted volumes and capacities, preserved flows and evidence of decreased gas exchange. High-resolution computed axial tomography demonstrates evidence of fibrosis and lung remodelling such as honeycomb cysts and traction bronchiectasis. There is no known effective treatment for IPF, but lung transplantation improves survival.
Idiopathic pulmonary fibrosis (IPF) is the most common of the idiopathic interstitial pneumonias and the one that is unresponsive to treatment.1,2,3,4 The purpose of this review is to help clinicians understand the disease and appropriately investigate suspected cases. IPF, known in Europe as cryptogenic fibrosing alveolitis, is a clinical term that describes a chronic fibrosing interstitial pneumonia with no known cause.1,2,3,4 In the past the term IPF included a number of idiopathic interstitial pneumonias that, in the revised histological classification, are now considered separate clinicopathogical entities. There are 7 distinct histological groups (see Table 1) with similar clinical presentations, but their clinical courses and responses to therapy differ.4,5,6,7,8,9,10,11,12
Table 1.
Epidemiology
The incidence and prevalence of IPF have been difficult to determine because until recently the diagnostic criteria were uncertain and the terminology was poorly defined.3,4 The General Practice Research Database and the 4th Morbidity Survey in General Practice from the United Kingdom suggest a prevalence of IPF of 1.5–1.8 per 10 000 person-years and an incidence of 0.5 per 10 000 person-years.13 IPF is more prevalent than other interstitial pneumonias and is diagnosed more frequently in men (male: female prevalence ratio 1.4:1.0; male:female incidence ratio 1.3:1.0).14 IPF is most commonly seen in patients between the ages of 40 and 70, and the majority of patients are over 60 years of age.3,6,7,8,9,11 The mean length of survival after diagnosis is 3.2–5 years.1,2,3,4,5,6,7,8,9
Etiology
Although the pathogenesis of IPF remains elusive, a number of conditions and risk factors are associated with the disease. Cigarette smoking increases the risk of IPF, with an odds ratio (OR) of 2.3 (confidence interval [CI] 1.3–3.8) among smokers with 20–40 pack-years of smoking.15 Several occupational factors, adjusted for age and smoking, are associated with IPF, such as farming (OR 1.6, 95% CI 1.0–2.5), hairdressing (OR 4.4, 95% CI 1.2–16.3), stonecutting (OR 3.9, 95% CI 1.2–12.7), and exposure to livestock (OR 2.7, 95% CI 1.3–5.5), birds (OR 4.7, 95% CI 1.6–14.1) and dust from metals (OR 2.0, 95% CI 1.0–4.0) and vegetables (OR 4.7, 95% CI 2.1–10.4).16,17 Although viruses have not been clearly implicated in the pathogenesis of IPF, several viral proteins and antibodies to viruses are associated with the disease, such as the Epstein–Barr virus,18 influenza A virus,19 hepatitis C virus,20 parainfluenza viruses 1 and 3,21 HIV-121 and herpesvirus 6,21 to name a few. Some people may have a genetic predisposition to IPF, as there have been cases of families in which at least 2 primary members had IPF.22 Familial IPF may be inherited as an autosomal dominant trait with variable penetrance, but this speculation has not been supported in each instance.4,23,24,25,26 Inherited abnormalities in surfactant proteins24 and the interleukin-1 (IL-1) receptor antagonist,25 as well as polymorphism of the tumour necrosis factor-alpha gene25 and complement receptor 1 gene,26 have been associated with some cases of IPF, which suggests that obscure biochemical aberrancies can lead to an IPF-like condition.24,25,26
Pathogenesis
Despite some of these associations, IPF has traditionally been thought to occur as a result of an initial injury to the lung that causes the recruitment of inflammatory cells, release of cytokines and eventually, from increased fibroblast activity, parenchymal remodelling and fibrosis.27,28,29,30,31,32,33 However, pulmonary fibrosis has been demonstrated in animal models in the absence of inflammatory cells.33,34 Furthermore, because inflammatory suppressive agents do not seem to be effective, it may be that IPF is not caused by inflammatory cells but that inflammatory cells are secondarily involved.29,30,31,32,33
More recent research has demonstrated that alveolar epithelial cells, as a consequence of injury by an unknown cause, may be the source of a number of fibrogenic cytokines such as transforming growth factor-beta 1,33,34,35,36,37,38 platelet-derived growth factor,39 tumour necrosis factor-alpha,40,41 IL-1,40 insulin-like growth factor142 and basic fibroblast growth factor.43 Release of these cytokines may result in fibroblast proliferation and migration to various sites in the lung, followed by differentiation of the fibroblast phenotype.44,45 This differentiation of the fibroblast is likely the key to the chronic nature of IPF. First, the differentiated cell seems to be more resistant to apoptosis (natural cell death), a process that is important in the repair of tissue without excessive scarring;45,46 second, the altered cells demonstrate a heightened responsiveness to fibrogenic cytokines such as transforming growth factor-beta and basic fibroblast growth factor.45,46 These events would lead to prolonged retention of fibroblasts, continued connective tissue protein synthesis and the formation of fibroblast foci, a histological hallmark in IPF.1,2,3,4
Attempts to elucidate an immunological basis for the pathogenesis of IPF have been made. In brief, it appears that the type 2 T-cell response predominates in IPF, with an increase in IL-4 and IL-13.47 Although levels of circulating autoantibodies such as antinuclear antibodies, anti-DNA antibodies, anti-cytokeratin 8 antibodies and immune complexes may be elevated,48 the significance of these abnormalities is unclear. In summary, although the histological features of IPF suggest an inflammatory or immunologic cause, no distinct pathophysiologic process has been identified.
Pathology
The pathological criteria for the diagnosis of IPF have undergone substantial changes. Under the current classification IPF refers to the clinical disease, and the typical histological features of IPF are called usual interstitial pneumonia.1,2,3,4 Usual interstitial pneumonia is characterized by temporal heterogeneity and the presence of fibroblast foci (Fig. 1D)1,2,3,4,9,10,11,12 and may be seen with other conditions such as pulmonary fibrosis associated with asbestosis, collagen vascular disease, chronic drug toxicity, chronic hypersensitivity pneumonitis and familial idiopathic pulmonary fibrosis, as well as in rare conditions such as Hermansky–Pudlak syndrome.3 However, these conditions are usually easily distinguished from IPF on clinical grounds.

Fig. 1: Gross and histological changes of idiopathic pulmonary fibrosis. A: Section of a lung removed at autopsy from a patient with IPF demonstrates honeycomb cysts. B: Enlargement of the area identified in 1A at a higher magnification shows the cystic lesions of honeycomb cysts. C: Whole-lung thin section highlights the subpleural and basal predominance of pathological changes of IPF, with less involvement of the lung tissue in the more central zones. The pleura is identified by (1); (2) is the preferred site of a diagnostic biopsy, at an interface between honeycomb lung and less involved lung tissue; (3) is an area of honeycomb lung; (4) is a more central area of the lung with minimal changes and appears grossly normal. D: Temporal heterogeneity of histological findings in a single biopsy observed to contain normal lung tissue (N), fibroblast foci (arrows) and interstitial fibrosis (*). Magnification is х40.
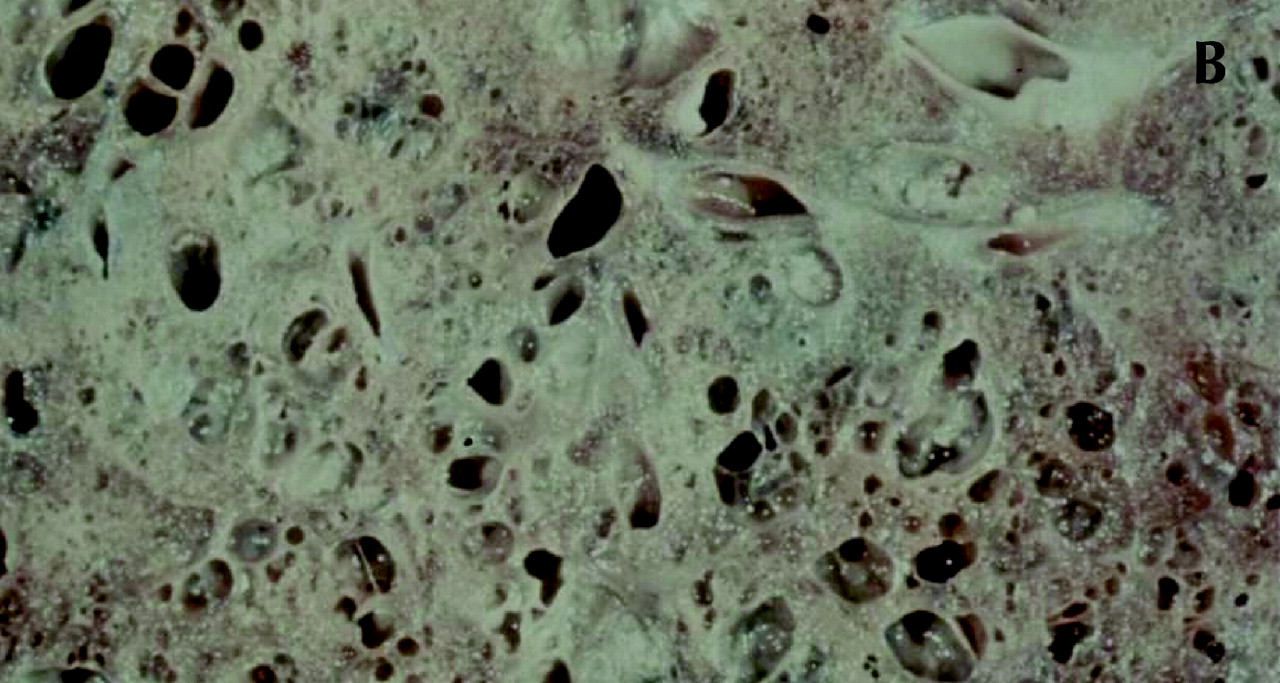
Figure 1. Continued.

Figure 1. Continued.

Figure 1. Continued.
The histological lesions of IPF are best seen with low-power microscopy, which demonstrates areas of normal-appearing lung tissue interspersed with interstitial fibrosis, inflammatory cells and distortion of the normal lung architecture to form honeycomb cysts and fibroblast foci (Fig. 1).1,2,3,4,9,10,11,12 Honeycomb cysts are enlarged and distorted airspaces resulting from destruction of the normal alveoli.4 Fibroblast foci are areas with palisades of fibroblasts and connective tissue located just beneath hyperplastic type 2 pneumocytes.1,4 The distribution of pathological changes is subpleural, paraseptal and prominent at the bases (Fig. 1).4,49,50
Under the current classification, other idiopathic interstitial pneumonias are termed nonspecific interstitial pneumonia, organizing pneumonia, diffuse alveolar damage, respiratory bronchiolitis, desquamative interstitial pneumonia and lymphoid interstitial pneumonia (Table 1).4 Lung diseases associated with these other idiopathic interstitial pneumonia patterns are all responsive to corticosteroid treatment.1,2,3,6,7,8,9,10,11,12 The histological findings of some lung biopsies may demonstrate more than 1 disease (e.g., lesions compatible with both nonspecific interstitial pneumonia and usual interstitial pneumonia). The prognosis is determined by the extent of changes compatible with usual interstitial pneumonia.2,9,10,11
Clinical features
Idiopathic interstitial pneumonias as a group generally present with cough and minimal or no sputum production. The most common clinical presentation of IPF is that of insidious progressive shortness of breath, or dyspnea, being present for at least 3 to 4 months.3,4,51 Cough may be present with or without sputum.3,4,51 On physical examination 25%–50% of patients with IPF have evidence of clubbing.3,4,5,51 The presence of clubbing is helpful in suggesting a diagnosis of IPF, but it is also observed in patients who have pulmonary fibrosis associated with rheumatoid arthritis, asbestosis or fibrosing nonspecific interstitial pneumonia.4,51 On auscultation, there are fine inspiratory crackles, heard best in mid- to end inspiration.3,4,51 With more severe disease, increased right heart pressure and right ventricular failure may be evident.4,52
The clinical diagnostic approach begins with a detailed history, physical examination, radiological imaging, lung function studies, blood tests and tissue analysis.3,4,51 The history should include a detailed occupational history and any exposures to the potential causative agents mentioned earlier. Since the majority of patients with IPF are over 60 years, a patient younger than 50 years with the clinical and radiological features of IPF may well have another disorder. Similarly, since IPF is confined to the lungs, the presence of systemic complaints suggests an alternate diagnosis or a concomitant illness such as an infection.
Investigations
Imaging
A plain film of the lungs usually demonstrates reticular abnormalities and honeycombing, especially at the bases.3,4,49,50,51,53 These findings, however, are not specific to IPF. High-resolution CT scanning has greatly improved the ability to visualize and characterize the abnormalities in IPF.3,6,7,8,13,49,50,53 Changes felt to be secondary to IPF on scanning include reticular densities, traction bronchiectasis, honeycomb cysts, septal thickening and a subpleural distribution of the abnormalities described earlier (Fig. 2).3,8,9,49,50,53 Again, these changes are most prominently seen at the bases.3,49,50,53 The presence of ground-glass appearance on high resolution CT is not characteristic of IPF.3,49,50,53 IPF can be correctly diagnosed by CT scan in up to 80% of patients with biopsy-proven IPF.54,55 MRI scanning56 and positron emission tomography57 are currently under investigation for their accuracy in diagnosing IPF. Lung scanning with gallium, indium and technetium are of little value in diagnosing or following the progress of IPF.3
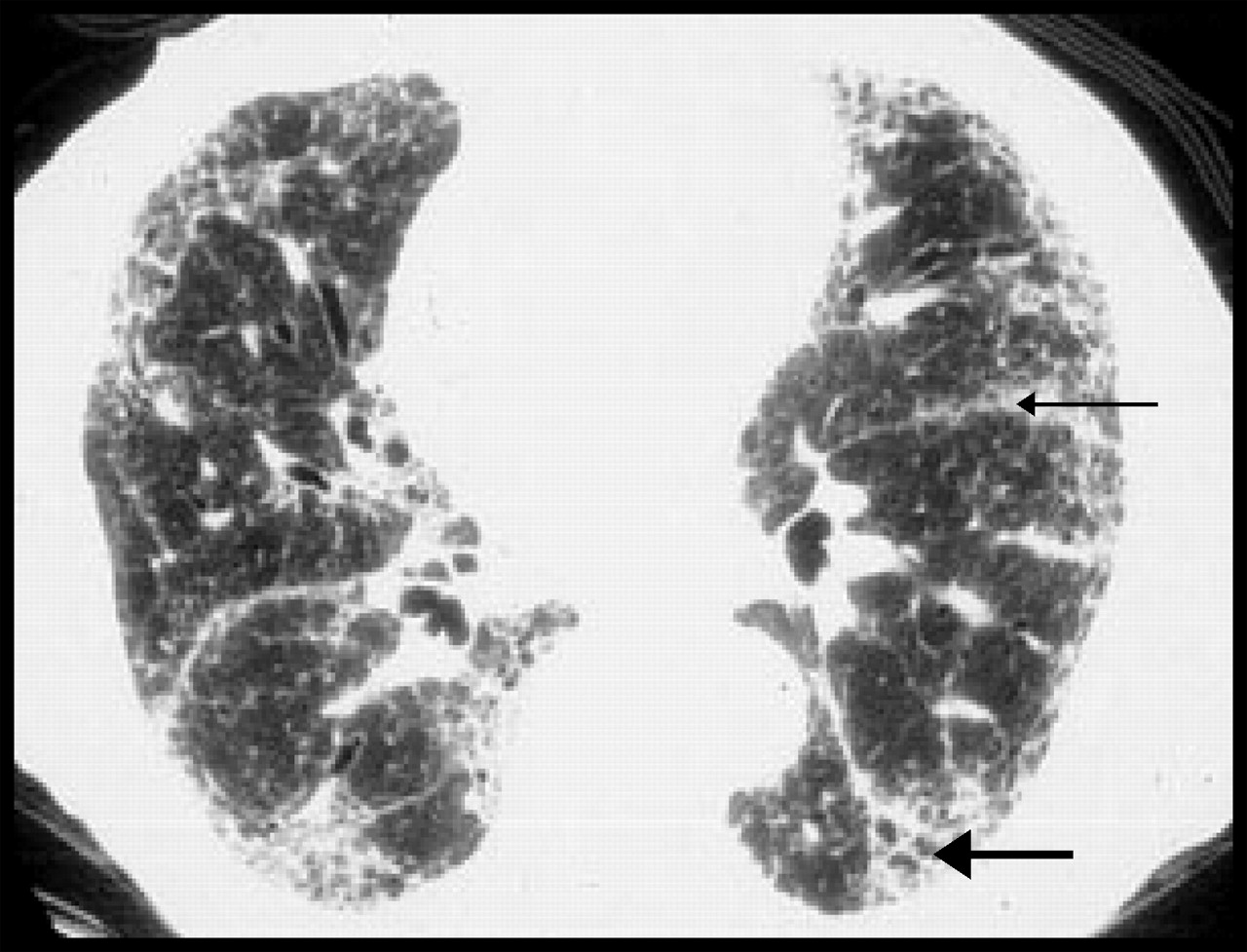
Fig. 2: Radiological changes of IPF on high-resolution CT. Subpleural fibrosis, honeycomb cysts, traction bronchiectasis (thick arrow) and paraseptal fibrosis (thin arrow) are apparent. Relatively minor changes are seen in the central portion of the lung.
Pulmonary function tests
Pulmonary function tests demonstrate the severity of disease and may explain the impact on function and exercise limitation.58 Pulmonary function studies have shown restrictive lung volumes and capacities, proportionately normal or supernormal flow relative to the lung volumes and a decrease in the carbon monoxide transfer factor.6,7,8,58 There may be hypoxemia with a widened alveolar–arterial oxygen gradient that increases with exercise.58 Gas transfer and desaturation on exercise are well correlated with the extent of disease on high-resolution CT.6,7,8,58 To follow disease progression, serial measurements of vital capacity and forced vital capacity may be helpful and are noninvasive and inexpensive.6,7,8
Blood tests
Since IPF is isolated to the lung and by definition has no associated systemic disease, results of blood tests are generally normal. However, some people may have an elevated neutrophil count caused by a concomitant infection.3 Hypergammaglobulinemia is seen in some IPF patients;59,60 elevated rheumatoid factor and antinuclear antibody titres are seen in about 40% of patients with IPF.3,4,59,60
Tissue diagnosis
Bronchoalveolar lavage is an excellent research tool,37 but it does not help in diagnosis or in following patients with IPF for either their response to therapy or the progression of disease.61,62 Because the diagnosis of usual interstitial pneumonia requires a spectrum of histological changes per high-power field, transbronchial biopsy, which provides a very small sample, has limited use as a diagnostic tool. However, bronchoalveolar lavage and transbronchial biopsy can help rule out other causes of pulmonary interstitial infiltrates such as sarcoidosis, hypersensitivity pneumonitis, malignant disease or infection.63
Surgical lung biopsy remains the “gold standard” for diagnosis. It is, however, by no means always definitive: the size of specimens, sites of biopsy used, expertise of pathologists and interobserver differences among pathologists are factors that may preclude a conclusive diagnosis.1,2,3,4,9,10,11,12 The site of the biopsy should be chosen on the basis of high-resolution CT findings9,35,37,64 and ideally be at the interface of involved and less involved lung tissue in order for the biopsy to show the pathological process at an active and recognizable stage.9 A biopsy of more than 1 site in the lung is also helpful in diagnosis of both IPF and other interstitial lung diseases.8,64 Indications for a surgical lung biopsy suggested by the American Thoracic Society and European Respiratory Society4 and by Raghu and colleagues55 are listed in Box 1.
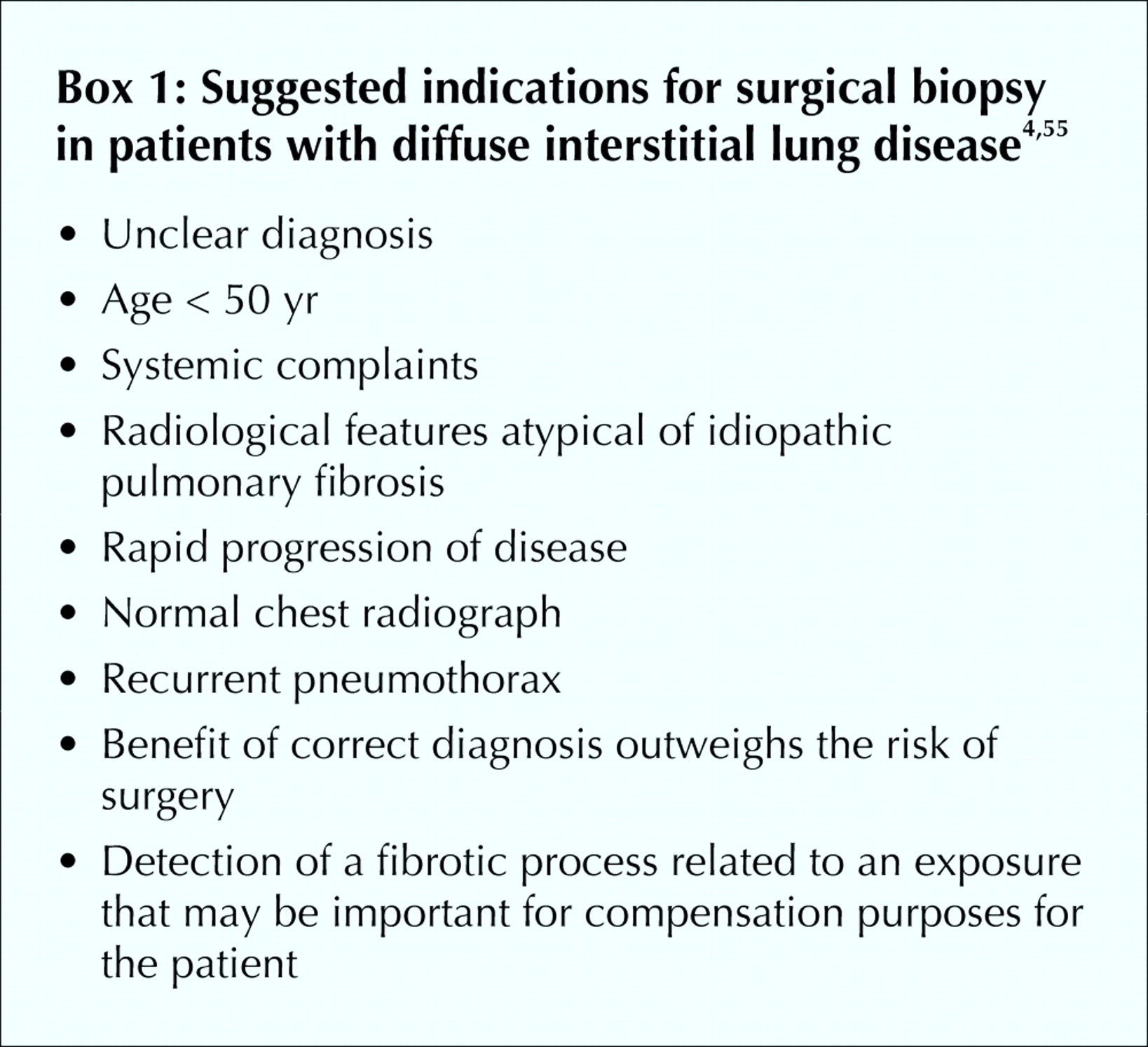
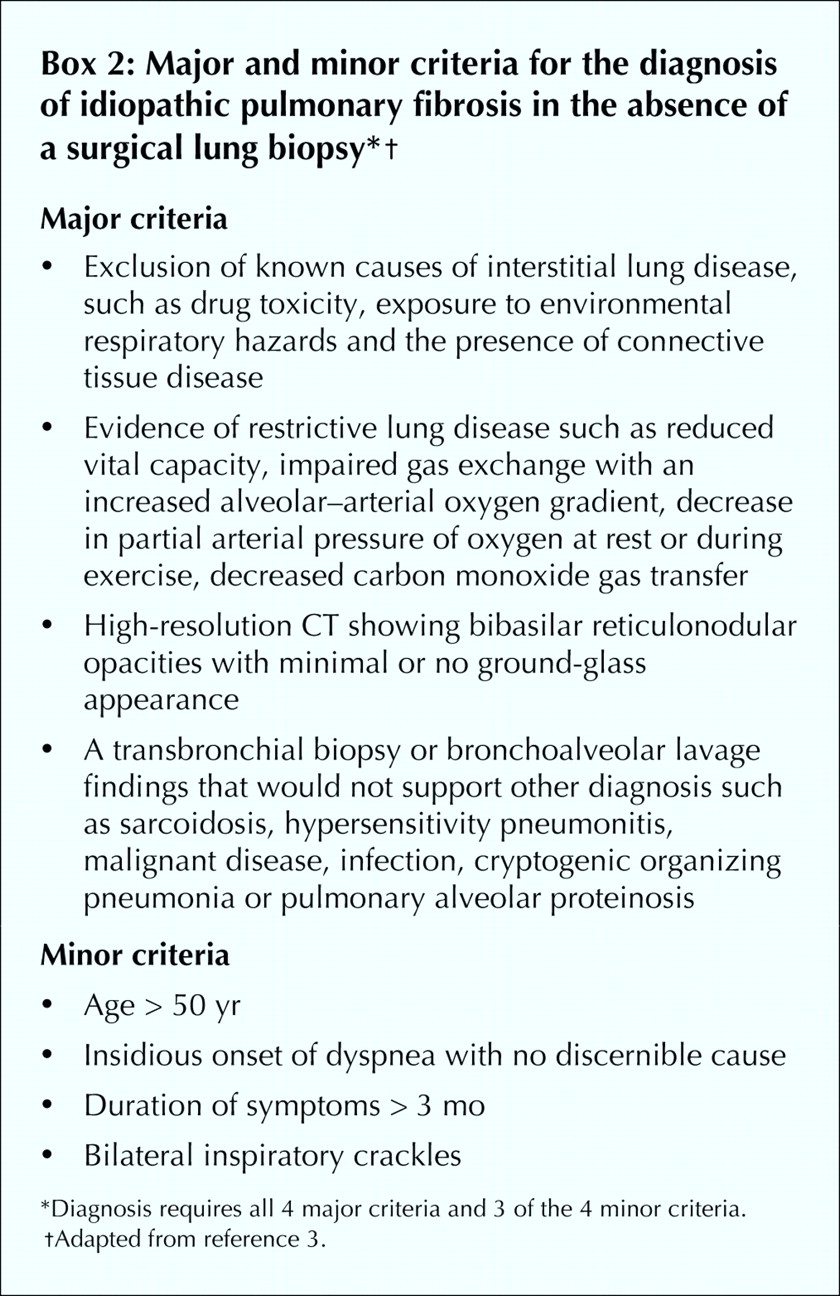
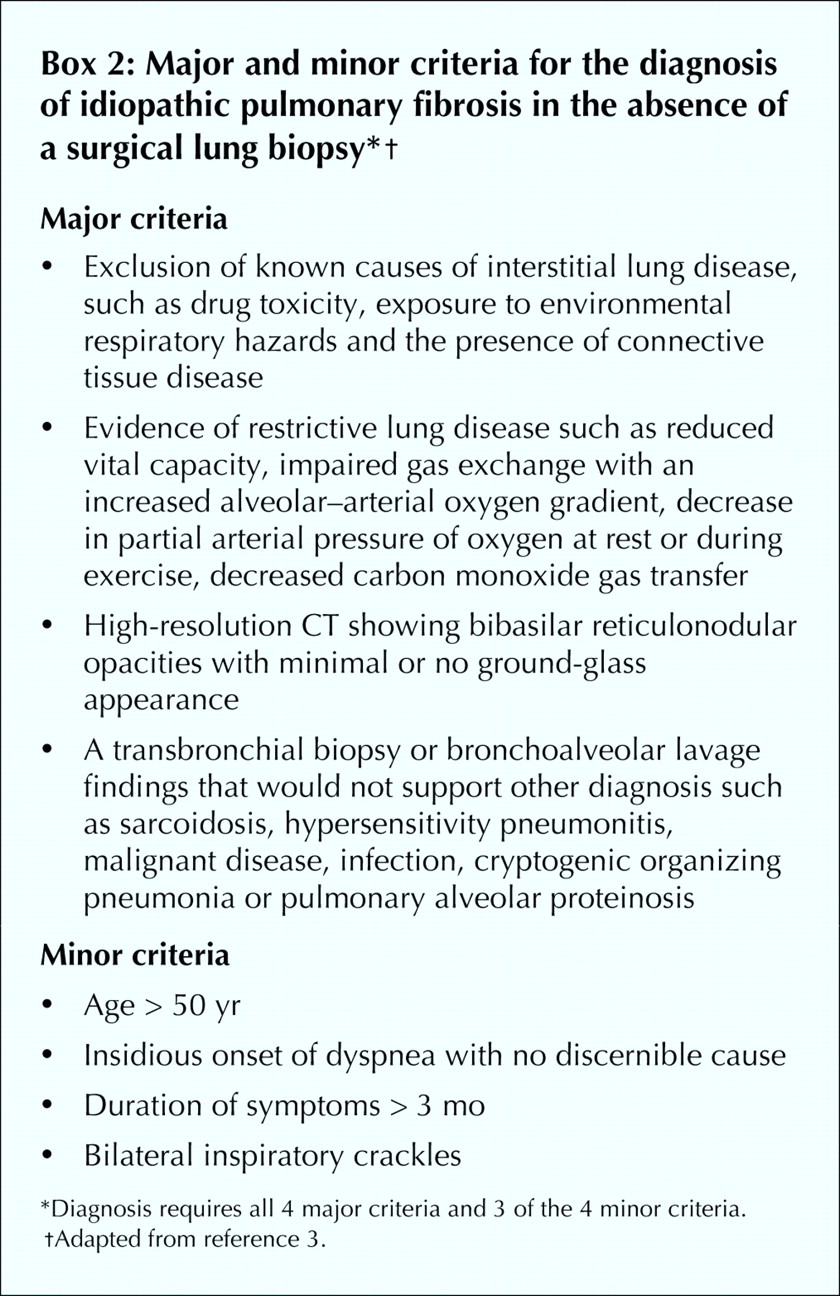
When patients have systemic complaints, a lung biopsy is suggested to rule out other causes of interstitial lung disease.4,55 If a radiological feature is not characteristic of IPF (e.g., if it is not subpleural, paraseptal or predominant at the bases), then a lung biopsy is indicated.4,55 The decision for a surgical lung biopsy has to be carefully considered, as in one series of biopsies, 10 of 60 patients with IPF who had received an “uncertain diagnosis” or were experiencing a “rapid decline” died within 1 month of the biopsy.65 No specific or consistent clinical feature distinguished patients with a poor prognosis after biopsy from the others. In the absence of a surgical lung biopsy the presence of 4 major and 3 of 4 minor diagnostic criteria assures the likelihood of a correct diagnosis of IPF (Box 2).4,52,53,66
Management
The management of IPF requires a methodical approach, regular evaluations and implementation of pharmacological and nonpharmacological modalities. Pharmacotherapy consists of immunosuppressive agents or antifibrotic agents; many drug therapies have both functions.
Immunosuppressive therapy
Currently no single or combined immunosuppressive agent has been found to improve survival of patients with IPF.4,30,31,32 Although corticosteroids have been standard therapy for years,30,32 no evidence of benefit in survival has been established. The use of corticosteroids is also associated with side effects and complications.30,32,67 Earlier reports demonstrating improvement with corticosteroids3,51 are difficult to interpret because, given the confusion of diagnostic criteria, some patients who responded may have had another disorder.4,9,10,11,12,68 For example, the re-examination of surgical lung biopsies in one centre yielded a number of cases that would now be called nonspecific interstitial pneumonia, which is steroid responsive.68 Despite the lack of evidence, a common therapy currently used for IPF is corticosteroids in conjunction with azathioprine.30,31,32,69 The combination of prednisone (20 mg/d) and azathioprine (3 mg/kg daily but not to exceed 200 mg/d) was reported to tend to stabilize lung function, but no comment on mortality rates could be made since the duration of the study was only 1 year.69 Before starting the combined therapy it may be wise to determine if the patient has latent tuberculosis by doing a tuberculin skin test.70 Patients taking azathioprine should be monitored for hepatotoxic effects, renal dysfunction and bone marrow suppression.69 Patients taking steroids should be assessed for side effects and complications of chronic corticosteroid use, and osteoporosis prevention with vitamin D and calcium supplementation and bisphosphonates should be undertaken.32,67 Corticosteroids combined with cyclophosphamide have been less successful in stabilizing lung function or affecting mortality.31
Antifibrotic therapy
A number of agents that interfere with collagen synthesis have been tested in clinical trials: pirfenidone, interferon gamma-1b, interferon beta-1a, colchicine and penicillamine.71 Many of the trials used small numbers of patients, and although some patients demonstrated improved or stabilized lung function, no antifibrotic agent has had any effect on mortality.
Pirfenidone is a pyridone molecule that inhibits connective tissue synthesis in an animal model of pulmonary fibrosis.72 In one study, 54 patients who had failed other therapeutic measures were given pirfenidone on a compassionate basis for 1 year.73 From the time of entry to follow-up at 24 months, there was no deterioration in the percent of predicted forced vital capacity, total lung capacity, gas transfer, oxygen saturation and litres of supplemental oxygen used. A study from Japan using pirfenidone drew a similar conclusion.74
Interferon gamma-1b is a glycoprotein found to inhibit collagen synthesis by fibroblasts and in animal models of pulmonary fibrosis.75 In a randomized trial 9 patients received interferon gamma-1b (200 μg/d 3 times per week) plus prednisolone (7.5 mg orally per day) and were compared with 9 patients who received prednisolone (7.5 mg orally per day) alone.76 All 9 patients receiving interferon gamma-1b plus prednisolone had an improvement in their lung function. A larger, multicentred, double-blind randomized trial evaluating the survival benefit for patients taking interferon gamma-1b plus prednisone versus those taking prednisone and placebo revealed that interferon gamma-1b did not affect progression of the disease, lung function or quality of life.77 Survival benefits could not be adequately assessed because the duration of the study was 1 year.
Interferon beta-1a has been reported to inhibit proliferation of fibroblasts and collagen synthesis by fibroblasts and also to inhibit collagen lattice contraction.78 A randomized placebo-controlled trial involving 167 patients did not demonstrate that interferon beta-1a was of therapeutic benefit.79
Colchicine inhibits fibroblast proliferation and collagen deposition80,81 and decreases the expression of platelet-derived growth factor-beta,a mesenchymal mitogen.80,81Studies in which colchicine was administered to patients with IPF have failed to demonstrate a survival advantage.80,81
Penicillamine is a chelating agent that suppresses inflammation and collagen deposition.82 Small trials investigating the use of penicillamine in patients with IPF have failed to demonstrate clinical efficacy or survival benefits.81
Novel antifibrotic treatments are currently being studied (Table 2).83,84,85,86,87,88,89
Table 2.
Nonpharmacological treatment
In a study by Lok, patients with IPF were found to do better when followed in an interstitial lung disease clinic.90 In that context, attention to fitness, good nutrition, oxygen supplementation when necessary, treatment of concomitant illness, prevention of morbidity from therapy and early referral for transplantation are all likely to be important in the reduction of morbidity in and management of IPF.90,91 Lung transplantation has been shown to improve survival in patients with IPF.92,93 In one study a single lung transplantation reduced the risk of death by 75% (95% CI 8%–86%, p = 0.03) compared with patients with IPF on the transplant waiting list.92No cases of disease recurrence were reported in the donor lung after transplantation. Since IPF is a progressive disease and no treatment is known to prolong survival other than lung transplantation,92,93 patients should be referred for transplantation assessment soon after diagnosis.
Conclusion
IPF is a progressive interstitial lung disease with no known cause or effective treatment. Combinations of existing therapies may be found to be effective when studied in future clinical trials. Currently, though, patients with IPF require both a comprehensive approach to management, ideally from specialized clinics, and early referral for transplantation.
Footnotes
-
This article has been peer reviewed.
Contributors: Nasreen Khalil was the principal author and contributed substantially to the analysis and review of the literature and to composing the manuscript. Robert O'Connor contributed equally to the composition of the review article. Both authors gave final approval of the version to be published.
Competing interests: None declared.
Correspondence to: Dr. Nasreen Khalil, Associate Professor of Medicine, Jack Bell Research Centre, 2660 Oak St., Vancouver BC V6H 3Z6; fax 604 875-4497; nkhalil{at}interchange.ubc.ca
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
In this issue
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools






Jump to section
Related Articles
Cited By...
- All-trans-retinoic acid inhibition of transforming growth factor-{beta}-induced collagen gel contraction mediated by human Tenon fibroblasts: role of matrix metalloproteinases
- Association of fine specificity and repertoire expansion of anticitrullinated peptide antibodies with rheumatoid arthritis associated interstitial lung disease
- Lymphangiogenesis as a Prerequisite in the Pathogenesis of Lung Fibrosis
- MMP2 Activity is Critical for TGF{beta}2-Induced Matrix Contraction--Implications for Fibrosis
- Naringenin: A Potential Immunomodulator for Inhibiting Lung Fibrosis and Metastasis
- Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis
- Activation of Discoidin Domain Receptor 1 on CD14-Positive Bronchoalveolar Lavage Fluid Cells Induces Chemokine Production in Idiopathic Pulmonary Fibrosis
More in this TOC Section
Similar Articles

