An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation
by
 , , and
, , and
Sarmistha Saha
1,*, 
Brigitta Buttari
1 ,
,
Emiliano Panieri
2,
Elisabetta Profumo
1 and
Luciano Saso
2
1
Department of Cardiovascular, Endocrine-Metabolic Diseases and Aging, Italian National Institute of Health, 00161 Rome, Italy
2
Department of Physiology and Pharmacology “Vittorio Erspamer”, Sapienza University of Rome, 00161 Rome, Italy
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(22), 5474; https://doi.org/10.3390/molecules25225474
Submission received: 19 October 2020
/
Revised: 13 November 2020
/
Accepted: 19 November 2020
/
Published: 23 November 2020
(This article belongs to the Special Issue Chemistry, Biology and Pharmacology of Modulators of Oxidative Stress)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Inflammation is a key driver in many pathological conditions such as allergy, cancer, Alzheimer’s disease, and many others, and the current state of available drugs prompted researchers to explore new therapeutic targets. In this context, accumulating evidence indicates that the transcription factor Nrf2 plays a pivotal role controlling the expression of antioxidant genes that ultimately exert anti-inflammatory functions. Nrf2 and its principal negative regulator, the E3 ligase adaptor Kelch-like ECH- associated protein 1 (Keap1), play a central role in the maintenance of intracellular redox homeostasis and regulation of inflammation. Interestingly, Nrf2 is proved to contribute to the regulation of the heme oxygenase-1 (HO-1) axis, which is a potent anti-inflammatory target. Recent studies showed a connection between the Nrf2/antioxidant response element (ARE) system and the expression of inflammatory mediators, NF-κB pathway and macrophage metabolism. This suggests a new strategy for designing chemical agents as modulators of Nrf2 dependent pathways to target the immune response. Therefore, the present review will examine the relationship between Nrf2 signaling and the inflammation as well as possible approaches for the therapeutic modulation of this pathway.
1. Introduction
Inflammation is generally a pervasive response to disturbances in the tissue homeostasis due to a variety of stimuli such as pathogens, tissue injury, or contaminants which involves the activation of innate and adaptive immunity. During the last few decades, inflammation has been the focus of extensive research mainly aimed at elucidating its role in host defense mechanisms; however, at present, inflammation is linked to a wide range of diseases from allergy, cancer, Alzheimer’s disease, and many others. Thus, it is now one of the hottest topics in medical science.
The primary focus of inflammation is to remove the source of disturbance and restore the tissue homeostasis [1]. Traditionally, innate immunity is the rapid response in the form of phagocytosis, whereas the adaptive immunity is antigen-dependent and characterized by an immunological memory which enables mounting a more efficient immune response on subsequent exposure to the same antigen. The hallmark of this inflammatory response is the production of soluble signaling molecules called cytokines. Inflammation involves a cascade of complex events. The first step is the detection of an infection signal [2] and/or damaged tissues [3] which is mediated by the pathogen-associated molecular patterns (PAMPs) that specifically recognize the molecules expressed by the pathogens. Damage-associated molecular patterns (DAMPs), are endogenous molecules that recognize cells committed to death and activate the immune system via the interaction with pattern recognition receptors (PRRs). In response to the successful recognition of these signals, either transmembrane Toll-like receptors (TLRs) or the inflammasomes activate specific immune signaling pathways, resulting in the subsequent activation of nuclear factor kappa-light-chain-enhancer of activated B (NF-κB). As a consequence, NF-κB dissociates from IκB and translocates to the nucleus, where transcription process is up-regulated. These events further lead to the next stage of the cascade, which involves the secretion of pro-inflammatory cytokines, such as interleukin-1-beta (IL-1β), IL-6, tumor necrosis factor-alpha (TNF-α) and others [4]. These then recruit immune cells, such as monocytes and neutrophils at the site of injury resulting in the generation of reactive oxygen and nitrogen species (ROS, RNS) that damage macromolecules including proteins and DNA. In normal conditions, restoration blocks any further neutrophil recruitment for wound healing and reestablishes tissue homeostasis. In chronic “inflammation” cellular damage risk is multi-fold with sustained inflammatory response that causes tissue injury.
Inflammatory cells such as mast cells, macrophages, monocytes, and lymphocytes release inflammatory mediators including cytokines, chemokines, and prostaglandins which further recruit inflammatory cells to the site of injury resulting in a respiratory burst and elevated oxidative stress. These further recruit macrophages and directly activate NF-κB, Mitogen-Activated Protein Kinase (MAPK), and Janus kinase (JAK)- signal transducer and activator of transcription protein (JAK-STAT) signaling pathways associated with the inflammatory response [5,6]. The activation of transcription factors, such as NF-κB and Nrf2, are key components of inflammation signaling cascades and oxidative stress responses [5]. These transcription factors and their functional interrelation will be described in the following sections.
2. Nrf2-Keap1 Signaling Pathway
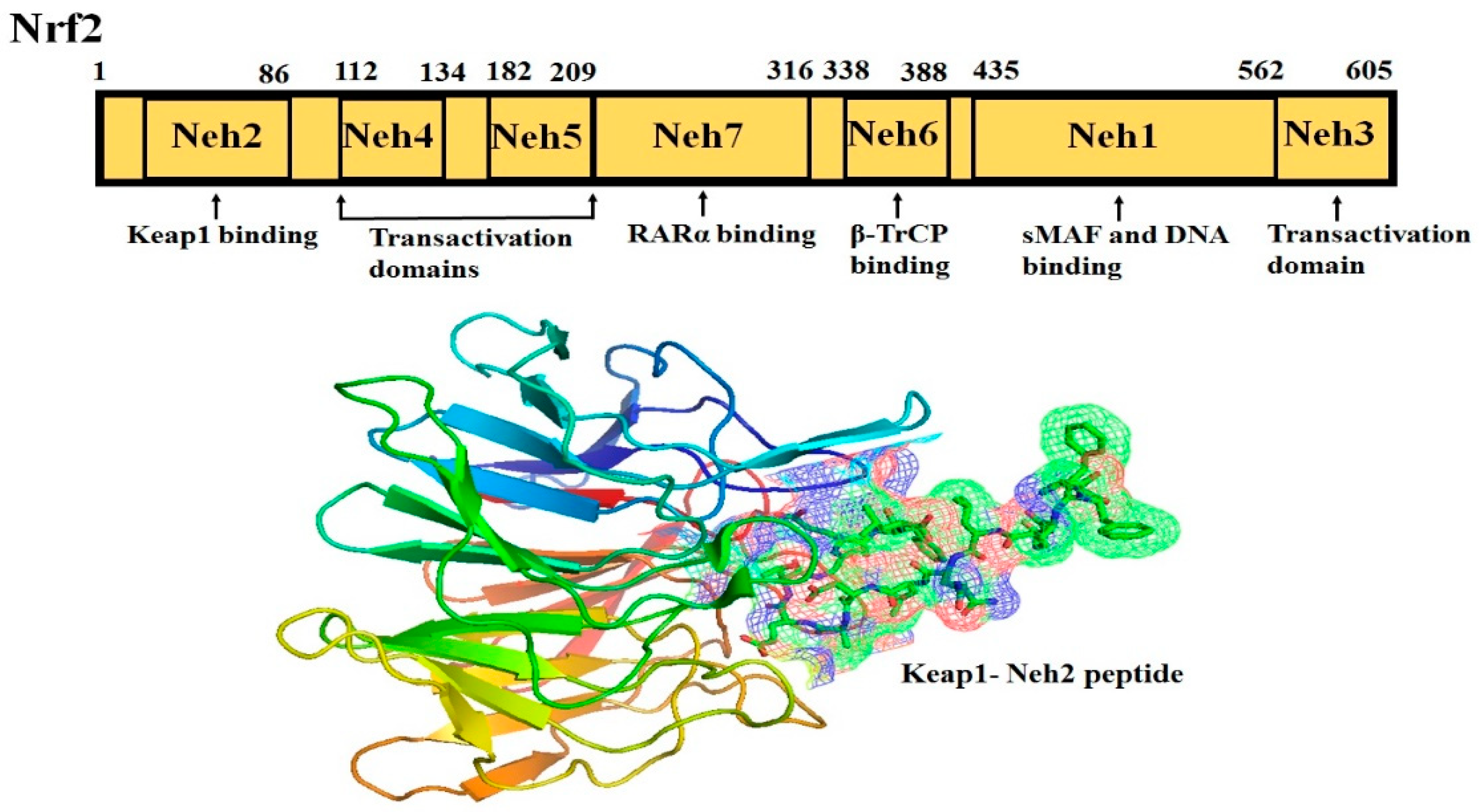
Nrf2 (NF-E2-related factor 2), a member of the Cap’n’collar (CNC) transcription factor family, consists of 605 amino acids and is divided into seven highly conserved functional domains, known as Neh1-Neh7 (Figure 1). The N-terminal domain influences the stability and ubiquitination of Nrf2 by its negative regulator Keap1, while the Neh5 domain is responsible for the cytoplasmic localization of Nrf2 [7]. The Neh1 domain has a cap ‘n’ collar basic-region leucine zipper (bZIP) domain, which regulates DNA-binding [8] and a nuclear localization signal (NLS) that is responsible for the nuclear translocation of Nrf2 [9]. The Neh3, Neh4, and Neh5 are transactivation domains mediating the interaction of Nrf2 with other coactivators [10,11]. Neh6 domain with serine-rich residues is a negative regulatory domain which binds to a β-transducin repeat-containing protein (β-TrCP) leading to Nrf2 ubiquitination [12]. The Neh7 domain inhibits Nrf2-ARE signaling pathway by promoting the binding of Nrf2 to the retinoic X receptor α (RXRα) [13].
The Neh2 domain, an N-terminal regulatory domain, contains seven lysine residues that influence the ubiquitin conjugation [14] and two peptide binding motifs (ETGE and DLG) that regulate Nrf2 stability by promoting its binding to different proteins [15]. The ETGE and DLG motifs interact with Keap1, which is a substrate adaptor protein for the Cullin 3- dependent E3 ubiquitin ligase complex that facilitates Nrf2 ubiquitination and its proteasomal degradation under normal physiological conditions [15,16,17].
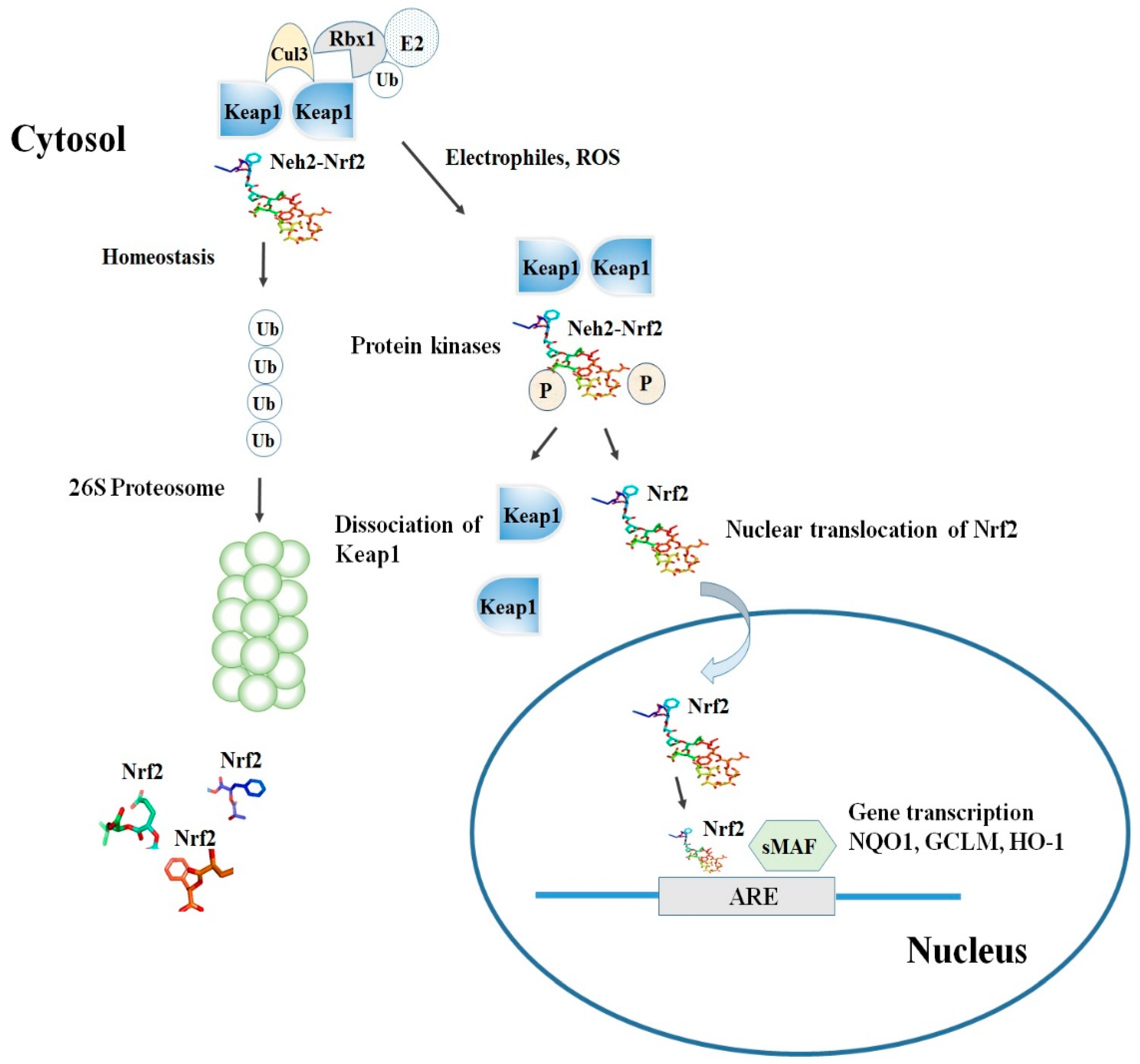
Keap1, consisting of 624 amino acids, is a cysteine-rich protein, containing 27 cysteine residues in humans [18]. Keap1 is divided into five domains, an N-terminal region (NTR), a Tramtrack and Bric-à-Brac (BTB) domain, a central intervening region (IVR) with a nuclear export signal (NES) mediating the cytoplasmic localization of Keap1 [19], six Kelch repeats, and a C-terminal domain (CTR) (Li et al., 2004). The BTB domain is responsible for Keap1 homodimerization and its binding to the cullin-based (Cul3) E3 ligase, leading to the formation of Keap1-Cul3-RBX1 (Ring box protein-1) E3 ligase complex [20], whereas Kelch repeats are thought to mediate the binding of Keap1 to Nrf2 and p62 (Figure 2) [21,22].
Among the others, the cysteine residues C151, C273, and C288 are highly reactive and susceptible of covalent modifications by ROS, RNS, H2S and other electrophiles. In this respect, the S-sulfenylation, the S-nitrosylation and the S-sulfhydration of these critical residues were shown to induce conformational changes of Keap1 that ultimately promote the dissociation of Nrf2 and its stabilization [23,24,25]. Although the exact mechanism of the Nrf2-Keap1 interaction is still unknown, two models were reported in the literature to explain the regulation of Nrf2 stability. The first, also known as the ‘‘hinge and latch’’ model, postulates that Keap1 interaction with the ETGE domain acts as a hinge while a weaker interaction with the DLG motif acts as a latch [26].
When specific thiol residues are modified by electrophiles, the DLG motif dissociates from Keap1 causing a disruption in the alignment of Nrf2 lysine residues that ultimately prevent its ubiquitination (Figure 2) [23,27]. Consequently, Nrf2 is released from Keap1-Cul3-RBX1 complex and translocates into the nucleus wherein it heterodimerizes with small Maf proteins (sMaf). After release, Nrf2 binds to EpRE in the presence of small Maf, and up-regulates electrophile response element (EpRE)-mediated transcription [28]. This further activates the transcription of a battery of genes containing an antioxidant response element (ARE) within their promoter region [27]. In addition, the carboxy-terminal domain of Neh3 can interact with the transcription coactivator, CHD6 (chromo-ATPase/helicase DNA-binding protein) [10] while Neh4 and Neh5 can interact with another transcriptional co-activator, CBP (cAMP-response- element-binding protein-binding protein) [11]. Finally, Neh4 and Neh5 can also bind to the nuclear cofactor RAC3/AIB1/SRC-3, further expanding the list of Nrf2 functional interactors, to promote the expression of Nrf2-targeted ARE genes [29]. Interestingly, as shown by several studies, thiol modifications of specifically C151 in the BTB domain might represent a stress sensing-mechanism that prevents the Keap1-Cul3 interactions and thus facilitates Nrf2 activation in response to adverse conditions [30].
The second model, also known as Keap1-independent regulation, postulates that in normal conditions, Neh6 domain binds to the DSGIS and DSAPGS motifs of the β-TrCP (Beta-transducin repeats-containing protein), which in turn is a substrate receptor for the Skp1-Cul1-Rbx1/Roc1 ubiquitin ligase complex that drivesNrf2 ubiquitination [31]. The phosphorylation of Nrf2 in the Neh6 domain by glycogen synthase kinase-3 regulates the recognition of Neh6 domain by β-TrCP [32].
3. Nrf2 and NF-ĸB Interplay in the Regulation of Cellular Redox Pathway
It is assumed that Nrf2 and NF-κB signaling pathways cooperate to maintain the physiological homeostasis of cellular redox status and to regulate the cellular response to stress and inflammation. However, the molecular mechanisms underlying this functional interaction appear to be cell type and tissue specific, and are still under elucidation [33]. However, the observation that Nrf2-knockout mice exhibit a strong down-regulation of phase II enzymes expression, suggests that the regulation of these genes is strictly dependent on Nrf2 [34]. In addition, other studies also indicate that Nrf2 system is essential for the expression of phase I and phase III xenobiotic transporters [35,36].
NF-ĸB is a complex protein system constituted by transcription factors that regulate the expression of genes influencing innate and adaptive immunity, inflammation, oxidative stress responses, and B-cell development. NF-kB activation involves both the canonical and non-canonical pathways. In the canonical pathway, NF-ĸB is sequestered as inactive form in the cytoplasm by its inhibitory proteins, including IĸB family members and other ankirin repeats-containing regulators. In presence of specific stimuli such as proinflammatory cytokines, PAMPs, oxidative stress and growth factors, the IĸB kinase complex gets activated and phosphorylates IĸB proteins, ultimately leading to their ubiquitination and proteasomal degradation. As a consequence, p50/RelA and p50/c-RelNF-κB dimers are free to translocate into the nucleus and transactivate multiple target genes. All NF-κB proteins contain a conserved Rel homology domain responsible for dimerization and DNA-binding [37]. NF-κB proteins can be classified into two groups based on the presence or absence of a transactivation domain. RelA (p65), RelB, and c-Rel contain transactivation domains, whereas p50 and p52 do not, and thus, they require heterodimerization with the Rel proteins to induce transcription [37].
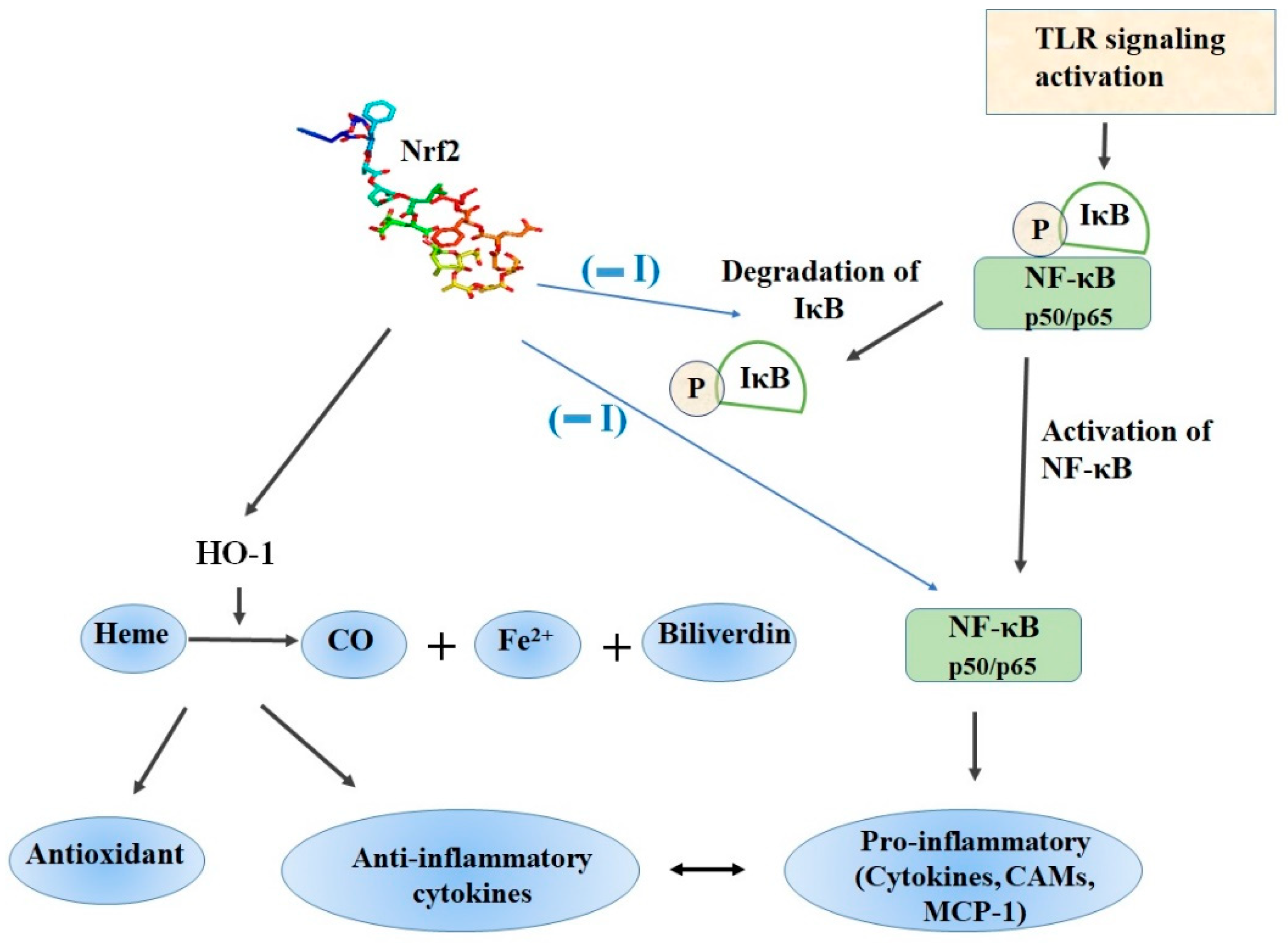
Nrf2/ARE signaling plays a crucial role in the protection against oxidative stress and is responsible for the maintenance of homeostasis and redox balance in cells and tissues [15]. In contrast, NF-κB is also a redox-regulated transcription factor, which regulates inflammatory responses and cellular injury [38]. After nuclear translocation, NF-κB induces the expression of proinflammatory cytokines (IL-1, IL-6, TNF-α), COX-2, iNOS, vascular adhesion molecules, and others (Figure 3) [38]. From a functional perspective, Nrf2 negatively controls the NF-κB signaling pathway by multiple mechanisms. Firstly, Nrf2 inhibits oxidative stress-mediated NF-κB activation by decreasing the intracellular ROS levels [39]. In addition, Nrf2 prevents the IκB-α proteasomal degradation and inhibits nuclear translocation of NF-κB [40]. Up-regulation of Nrf2 induces increase in the cellular HO-1 levels and subsequent increase in phase II enzymes expression blocks the degradation of IκB-α [41]. On the other hand, NF-κB decreases free CBP, which is a transcriptional co-activator of Nrf2 by competing with CH1-KIX domain of CBP while also promotes phosphorylation of p65 at Ser276 which in turn prevents CBP from binding to Nrf2 [42].
Accumulating evidence also suggests that Nrf2 counteracts the NF-κB-driven inflammatory response by competing with transcription co-activator cAMP response element (CREB) binding protein (CBP) [43,44,45]. The CBP-p300 complex is responsible for the acetylation of histones and exposes DNA for transcriptional machinery assembly. Apart from this, the CBP-p300 complex also acetylates lysine residues of non-histone proteins, including Nrf2 and p65 [8,42]. CBP was also reported to interact with Neh4 and Neh5 domains of Nrf2, leading to the acetylation of the Neh1 domain which is responsible for DNA-binding [42]. Since, CBP also preferentially interacts with phosphorylated p65 at Ser276, the overexpression of p65 limits the availability of CBP for Nrf2 interaction [42]. Accordingly, knockdown of p65 promotes Nrf2 complex formation with CBP [42]. Previous studies revealed that the transcription factor, NF-κB(p65) antagonizes the Nrf2-ARE signaling pathway by recruiting MafK-associated HDAC3 activity to the AREs-enriched HO-1 E1 enhancer and thereby deacetylates CBP suppressing its co-activator activity [46,47]. This event is also responsible for local “histone hypoacetylation” leading to repressed transcription and consequently, decreased induction of ARE-containing genes [48]. Thus, as a whole, Nrf2 transcription is down-regulated due to CBP sequestration as well as the abrogation of CBP co-activator activity caused by its deacetylation [46]. It was reported that the physical association of the N-terminal region of the p65 subunit of NF-κB with Keap1 can inhibit the Nrf-2 pathway [45]. However, numerous studies showed that different pathological sources of exogenous or endogenous stress such as lipopolysaccharide, oxidized LDL, pro-atherogenic oscillatory stress, smoke, shear stress, and other stress signals often activate both NF-κB and Nrf2-ARE signaling [49,50,51,52,53,54,55,56].
A recent paper studying inflammation in prostate cancer and castration-resistant prostate cancer showed that antioxidant and anti-inflammatory agents such as sulforaphane and curcumin can activate Nrf-2 and induces the nuclear accumulation of p120-Nrf1 which in turn significantly inhibits NF-κB DNA-binding activity by decreasing the transactivation of androgen receptor signaling [56].
Interestingly, some Nrf2 target genes such as NADPH quinone oxidoreductase I (NQO1), glutamate-cysteine ligase catalytic subunit (GCLC) and glutamate-cysteine ligase modifier subunit (GCLM) are also believed to possess NF-κB binding site. Thus, it is plausible that these two signaling pathways may cooperate in some specific cases, for example promoting chemoresistance of acute myeloid leukemia cells in response to bortezomib treatment [57,58]. This could be the reason compounds targeting NF-κB signaling conversely activate the Nrf2 pathway [59,60]. However, research focused on the potential impact of NQO1 on the NF-κB signaling led to conflicting results. One study suggested that the overexpression of NQO1 suppresses the expression of tumor necrosis factor (TNF)-α and IL-1 in LPS-stimulated THP-1 cells independently from NF-κB [61]. In contrast however, another study indicated that in NQO1 deficient mice NF-κB DNA-binding was inhibited in bone marrow, spleen, and thymus on LPS stimulation [62]. Similarly, nuclear thioredoxin (Trx) can modulate NF-κB activity via the reduction of cysteine residues on p50 and thus prevent DNA-binding [63,64,65,66] while cytoplasmic Trx prevents IκB degradation [67]. Cytoplasmic binding of Trx1 to IκB blocks its degradation and subsequently inhibits nuclear translocation of NF-κB, thereby decreasing its transcriptional activity [67]. However, the mechanism by which IκB is regulated by Trx1 was not yet elucidated.
Emerging evidence also revealed that NF-κB induction can promote Nrf2 activity via the small GTPase Ras-related C3 botulinum toxin substrate 1 (Rac1) protein (Cuadrado et al., 2014). For example, it was shown that the constitutively active form of Rac1 was able to induce the activation and the nuclear translocation of Nrf2ΔETGE-V5, which lacks four residues (ETGE) required for its recognition by Keap1, suggesting that Nrf2 induction through Rac1 actually occurs in a Keap1-independent manner (Cuadrado et al., 2014). In turn, Nrf2 suppresses Rac1-dependent activation of NF-ĸB pathway, whereas Nrf2 deficient cells promote NF-ĸB dependent inflammatory markers, suggesting the existence of a negative feedback mechanism that might regulate the intensity of Nrf2 activation under specific circumstances [68]. Indeed, LPS was shown to activate Rac1 which in turn up-regulates HO-1 expression through the NF-κB-dependent induction of Nrf2 signaling. Furthermore, the antioxidant enzyme HO-1, in turn, was found to suppress the NF-κB dependent inflammatory activity, indicating the crucial role of Nrf2 in the reduction of oxidative stress [68].
Some other reports stated that Nrf2 activation attenuated oxidative stress and neuronal inflammation through the suppression of the NOX4/ROS/NF-ĸB pathway [69,70]. Interestingly, Nrf2 overexpression modulates only targets lying downstream NF-κB in human aortic endothelial cells, since the expression of MCP-1, VCAM-1 and TNF-α was abrogated, without affecting NF-κB activity or IκB degradation [71,72,73,74].
Furthermore, sulforaphane, a potent Nrf2 inducer, was shown to prevent DNA-binding of NF-κB and to downmodulate the expression of NO, PGE2, and TNF-α in macrophages, while it failed to prevent LPS-induced IkB degradation or NF-kB nuclear translocation. Despite the role of NRF2 was not investigated, the authors proposed that sulforaphane might induce thiol modifications in the NF-kB subunits to impair its DNA-binding capacity [75]. In support of this evidence, several other reports showed enhanced NF-κB activation in Nrf2 knockout mice subdued to different stimuli such as LPS [76], ovalbumin [77], traumatic brain injury [78] and respiratory syncytial virus [79].
Nrf2 depletion in human monocytes reportedly enhanced TNF-induced inflammatory cytokines [80]. Mechanistically, TNF-induced sustained activation of Nrf2 and target genes expression through a TNFR1-dependent mechanism causing autocrine TNF production. The same study reported that Nrf2 silencing enhanced both TNF-α -induced proinflammatory genes expression as well as p50 and p65 DNA-binding, indicating a negative regulation of Nrf2 on NF-kB activation [80]. In agreement with these findings, disruption of Nrf2 signaling in knockout mice was shown to cause enhanced sensitivity to septic shock and lung inflammation in response to LPS or TNF-α. Accordingly, further analysis revealed that Nrf2-deficient MEFs (mouse embryonic fibroblasts) exhibited increased NF-kB activation in response to LPS and TNF-α treatment, suggesting that Nrf2 could suppress the inflammation induced by these stimuli blocking the induction of NF-kB [76].
In marked contrast; however, Nrf2 knockout in murine fibroblasts was shown to suppress p50 and p65 levels and enhance c-Rel protein content, whereas livers of Nrf2 knockout mice exhibited greatly reduced p65 levels. Based on this evidence the authors proposed that Nrf2 was actually required for the expression of NF-κB family members [81]. Taken together, these data point out the existence of a functional cross-talk between NF-kB and Nrf2 signaling while also highlight that this complex interplay can enhance or impair the inflammatory response, depending on the specific context and stimuli.
Oxidative stress has long been implicated in many pathological conditions and ROS accumulation plays a crucial role in the progression of inflammatory responses. Since, the mitochondrion is a primary site for ROS production under physiological and pathological conditions, a series of studies demonstrated the crucial role of the Nrf2 signaling pathway in cytosolic and mitochondrial ROS production [82]. For instance, NOX-2 (NADPH oxidase 2) activity is increased in Nrf2 deficient embryonic fibroblast cells and unrestricted Nrf2 activation in Keap1 knockout fibroblasts leads to increased NOX-4 activity [82]. In addition, up-regulated transcription of all the genes (GSTα2, NQO1, TrxR1, and GCLC) on ectopic Nox4 expression was observed to be ablated in cardiomyocytes of Nrf2-null mice, suggesting the essential role of Nrf2-Keap1 signaling in glutathione redox signaling [83]. A recent report shows that Nrf2 is associated with the outer mitochondrial membrane and protects mitochondria from oxidative stress, whereas this protective effect was absent in Nrf2 knockout mice [84]. Interestingly, Nrf2 was also recently found to protect mitochondria in response to oxidative stress suggesting a potential link between Nrf2 with mitochondrial outer membrane, despite, the exact molecular mechanisms through which Nrf2 interacts with the mitochondrial outer membrane and thus confers protection against oxidative insults, remains to be elucidated [84].
Nrf2-regulated target genes, NQO1 and NQO2 are two cytosolic flavoproteins catalyzing the two-electron mediated reduction of quinones to hydroquinones [85]. This catalytic reaction competes with the one-electron reduction reaction catalyzed by cytochrome P450 reductases, which generates highly reactive semiquinones [86]. Furthermore, NQO1 also maintains the reduced form of CoQ9 and CoQ10 inside the unilamellar or multilamellar vesicles, thereby protecting the plasma membrane from lipid peroxidation and free radicals. Knockout of NQO1 is associated with high susceptibility to immune disorders [87,88]. Moreover, the double knockout of both NQO1 and NQO2 strongly enhances the infiltration of neutrophils and macrophages in bronchial-associated lymphoid tissue, leading to an increase in pro-inflammatory cytokines in lung macrophages [89]. Another report showed that the induction of NQO1 by several synthetic triterpenoids resulted in the decreased expression of the pro-inflammatory inducible nitric oxide synthase (iNOS) in Hepa1c1c7 murine hepatoma cells, an effect that was strictly Nrf2-dependent and related to the direct interaction with Keap1 thiol groups [90].
Nrf2 directly modulates the ROS and RNS by regulating the levels of superoxide and peroxides through the induction of antioxidant enzymes such as superoxide dismutase, glutathione peroxidase and peroxiredoxin members [91,92,93]. In response to oxidative stress, Nrf2 translocates to the nucleus and activates the transcription of several antioxidant genes, such as glutathione biosynthetic enzymes (GCLM and GCLC), GSH-dependent antioxidant enzymes (glutathione peroxidase 2, GPX2), and glutathione S-transferases (GST) [94]. The Nrf2-dependent regulation of redox homeostasis operates at multiple levels of redox control, one of them involving the regeneration of reduced cofactors and protein-thiol groups from their oxidized counterparts. Well known examples include the conversion of glutathione disulphide (GSSG) into glutathione (GSH) by the glutathione reductase (GSR) [95], the reduction of oxidized thioredoxins by thioredoxin reductase (TrxR) [96] and Prx-SO2H by sulfiredoxin (Srx) [97]. Another mechanism, implies the transcriptional induction of enzymes involved in the generation of reducing factors, as occurs in the biosynthesis of GSH by glutamate-cysteine ligase (catalytic and regulatory) or NADPH by glucose-6-phosphate dehydrogenase (G6PDH) and other metabolic enzymes [98]. Accordingly, Nrf2-deficient mice show impaired expression of glutamate cysteine ligase (GCL) and strong depletion of the GSH levels [76]. As reported by Hartl et al., suppression of GSH, in turn, led to high susceptibility to oxidative stress, up-regulation of pro-inflammatory cytokines and impairment in T-cell responses [99]. Consistently, the overexpression of GCL was found to inhibit TNF-α-induced activation of NF-κB, AP-1, and JNK in rat hepatoma cells [100], while increase in GSH synthesis was shown to down-regulate the NF-κB activation and TNF-α release in LPS-stimulated alveolar type II (AT-II) epithelial cells [101]. Similarly, GSH was also found to down-regulate p38 activation as well as iNOS and COX-2 expression in rat peritoneal macrophages subdued to LPS stimulation [102], indicating that the GSH content can strongly influence the activity and function of molecular and cellular mediators of the inflammatory processes. This is in agreement with previous findings demonstrating that GSH-mediated redox status was indeed able to affect the Th1/Th2 balance while GSH depletion was found to inhibit Th1-associated cytokines release and favor Th2-associated responses [103,104].
In addition to this, several other mechanisms of Nrf2-mediated redox regulation were described. For example, the antioxidant protein thioredoxin (Trx) can be transactivated by hemin (ferriprotoporphyrin IX) through the binding of the Nrf2/small Maf complex to ARE [105]. The thioredoxin (Trx) system is a major intracellular thiol-reducing and ROS-scavenging pathway consisting of three components. Among them, the thioredoxin-interacting protein (TXNIP) acts as a negative regulator that binds to the reduced form of Trx and prevents it from reducing peroxiredoxin (Prx), thereby inhibiting Trx mediating function. In this context, Nrf2 was shown to inactivate the TXNIP expression and to suppress its induction under high glucose conditions through the interaction with ARE of the TXNIP promoter, which in turn inhibited the binding of MondoA to the carbohydrate response element in presence or absence of glucose [106]. This suggests the critical role of Nrf2 in redox regulation mediated by the TXNIP-Trx axis [106].
Another line of evidence revealed the key role of Nrf2 in cystine/glutamate exchange transport which ultimately regulates the intracellular GSH levels by influencing the uptake of cystine, the oxidized form of cysteine, a limiting substrate of GSH synthesis [107]. The cystine/glutamate exchange transport is found to be mediated by xc−system, which consists of two protein components, xCT and a heavy chain of 4F2 antigen [107]. Nrf2 was demonstrated to activate transcription of xCT in response to various stimuli, such as diethyl maleate (DEM), cadmium chloride, arsenite, and lipopolysaccharide, via the electrophile response element (EpRE) [108]. Electrophilic agents including DEM, are thought to promote the dissociation of Nrf2-Keap 1 complex and release of Nrf2 [108].
Another mechanism through which Nrf2 controls the redox balance is by influencing the expression of genes coding for proteins involved in iron storage, transport and metabolism. Iron and oxygen are interconnected and iron essentially acts as a cofactor in oxygen transport, oxidative phosphorylation, and metabolite oxidation reactions. However, excess iron leads to the development of oxygen-derived free radicals. Here, Nrf2 regulates iron metabolism pathway in response to oxidative and electrophilic stress by activating transcription of genes coding for ferritin heavy chain (Fth1) and ferritin light chain (Ftl) [109], thus increasing ferritin levels [110]. As a result, Nrf2 indirectly promotes iron storage and reduces the intracellular levels of redox-active free iron atoms. Moreover, Nrf2 also regulates labile iron (redox active, exchangeable and chelatable) by altering its transport across the cell membrane by up-regulating the expression of ferroportin (Fpn1) [111], which is a protein responsible for iron export from the cytosol to the extracellular milieu [112]. Similarly, Nrf2 plays a crucial role in the gene expression of metallothionein, a cysteine-rich metal-binding protein and another downstream target implied in the antioxidant pathway [113].
In response to the oxidative stress, cells induce another set of antioxidant enzyme including heme oxygenase-1 (HMOX1) and thioredoxin reductase-1 (TrxR1). Here, Nrf2 confers an additional layer of regulation for the protection against oxidative stress [114]. In the same study, oxidative stress triggered by hemin treatment-induced both proteasomal degradation and displacement of BACH1, a transcriptional repressor, from HMOX1 gene enhancers, which in turn allowed nuclear Nrf2 binding to the ARE motifs and subsequent transactivation of the HMOX1 gene [114]. In contrast, TrxR1 appeared to be directly regulated by Nrf2 without requiring previous BACH1 inactivation [114].
Since autophagy is reciprocally linked to oxidative stress, several studies also support the notion that Nrf2 can play a role in the regulation of autophagy. A crucial link between autophagy and Nrf2 is represented by p62, a ubiquitin-binding protein acting as an autophagy cargo receptor [115]. Under normal physiological conditions, p62 is constantly degraded during autophagy; however, under pathological oxidative stress conditions p62 accumulates and competitively binds to Keap1, thus causing the release of Nrf2 and the activation of its downstream target genes [116].
These findings hint that in response to oxidative stress, up-regulation of Nrf2 signaling activates a complex antioxidant response and maintains the redox homeostasis through the regulation of multiple mechanisms.
4. Cross-Talk between Nrf2 and Inflammation
Nrf2 is part of a multilayered network that plays a key role in redox homeostasis as well as in inflammation. Next, we will discuss the molecular mechanisms through which Nrf2 is regarded to influence both innate and adaptive immunity.
4.1. Role of Nrf2/HO-1 Axis in Inflammation
Heme oxygenase-1 (HO-1, HMOX1, EC 1.14.99.3), is an inducible 32 kDa protein and catalyzes the rate-limiting step of oxidative heme degradation. During this process, heme is converted into three bioactive products namely free iron, carbon monoxide (CO) and biliverdin, which is rapidly converted to bilirubin and plays crucial roles in inflammation, apoptosis and oxidative stress (Figure 3) [117]. Consistently with other antioxidant proteins, Nrf2 directly controls the expression of the HMOX1 gene codying for the HO-1 enzyme. The critical role of the Nrf2 mediated HO-1 expression for the anti-inflammatory activity was substantiated in a series of in vitro and in vivo experiments.
In a study focused on macrophage polarization, gene expression profiling showed that in vitro M-CSF-polarized macrophages were characterized by preferential expression of both CD163 and HO-1. Further analysis revealed that the experimental induction of HO-1 activity by cobalt protoporphyrin treatment resulted in the increased release of IL-10 in M2 macrophages, suggesting that the CD136/HO-1/IL-10 axis can influence the anti-inflammatory and regulatory functions of M2-macrophages [118]. In addition, the levels of CD206+ and HO-1 in M2 macrophages were found to correlate with the anti-inflammatory activity in diabetes-associated gastric pathology [119]. Similarly, hemin-mediated induction of HO-1 in macrophages contributed to anti-inflammatory activity in acute pancreatitis [120]. These findings implicated the therapeutic potential of targeted modulation of the heme-HO-1 system in macrophages.
After silencing Nrf2 via knock-out, the expression of COX-2, iNOS, IL-6, and TNF-α were dramatically increased in Parkinson’s disease [121]. Similarly, the Nrf2 activation led to a decrease in expressions of COX-2 and iNOS in vascular smooth muscle cells [122]. The enhanced expression of inflammatory proteins (COX-2 and iNOS,) and inflammatory cytokines (TNF-α and IL-6) was found to be accompanied by a concurrent increase in HO-1 and NQO1 content, implicating the role of Nrf2/HO-1 axis in inflammation. HO-1 can be rapidly induced by various types of oxidative/electrophilic stress which in turn can inhibit ROS and NO production in immune cells [123].
HO-1 knockout studies revealed a novel mechanism of the Nrf2/HO-1 axis in inflammation. For instance, HO-1 deficiency in human and mice enhanced systemic inflammation, impaired the fibrinolysis system and was accompanied by anemia, intravascular hemolysis and vascular endothelial injury [124,125,126]. Activation of Nrf2/HO-1 axis was found to reduce lipid peroxidation and TNF-α and IL-6 levels, and consequently to attenuate the inflammatory reaction in a tight junction dysfunction model [127]. Also, Taurine chloramine (TauCl), a product of activated neutrophils, was shown to induce the clearance of apoptotic cells by phagocytes, also known as efferocytosis, when injected into macrophages. Importantly, this process was abolished in Nrf2/HO-1 knockout macrophages, and in those cases, inflammation was resolved by Nrf2-mediated HO-1 up-regulation and subsequent production of CO [128]. Moreover, it was reported that Nrf2 mediated HO-1 up-regulation along with PPARγ ligands are responsible for the resolution of inflammation in chronic obstructive pulmonary disease [129].
Furthermore, the metabolites of heme degradation including CO and bilirubin also accelerate the resolution of oxidative stress and inflammation [117,130]. CO prevented bone destruction in collagen-induced arthritis and reduced cytokine levels in hind-limb ischemia-reperfusion injury [131,132]. Similar results were also obtained with bilirubin which showed amelioration of autoimmune encephalomyelitis [133], autoimmune hepatitis [134], endothelial activation and dysfunction [135]. Another study reported that bilirubin could exert a protective effect against endotoxic shock by preventing the expression of iNOS and thus the generation of NO [136]. Collectively, these findings suggest that Nrf2 largely mediates the anti-inflammatory action of HO-1.
Accumulating evidence indicates that the role of Nrf2/HO-1 signaling may be stimulus and cell type-specific [137,138]. It is already known that phosphorylation of Nrf2, Nrf2-Keap1 dissociation, and Nrf2/HO-1 signaling pathway is functionally interrelated (Figure 3). Indeed, a quite recent report showed that the Nrf2/HO-1 axis mediated the protective effect of isorhamnetin against H2O2 treatment in C2C12 myoblasts and that ERK (extracellular-signal-regulated-kinase) activation was required for the phosphorylation of Nrf2 and Nrf2/HO-1 signaling, whereas no involvement of PI3K, JNK, and p38 MAPK was observed [139]. In contrast, another report revealed that the MAPK-dependent phosphorylation of Nrf2 is required for its nuclear translocation in HepG2 hepatoma cells subdued to treatment with PDTC [140]. The authors demonstrated that the initial phosphorylation of Nrf2 on MAPK sites releases Nrf2 from Keap1 and this was followed by nuclear translocation and finally, DNA-binding [140]. Furthermore, nuclear translocation and DNA-binding also requires phosphorylation. Therefore, Erk-mediated phosphorylation of Nrf2 could be a prerequisite for its translocation despite the exact mechanism by underlying this process is yet to be elucidated.
4.2. Functional Interaction of Nrf2 with Inflammatory Mediators
It is well established that inflammation is a complex interplay of different inflammatory cells that release several signaling molecules such as arachidonic acid derivatives (leukotrienes and prostaglandins), phospholipid mediators (platelet-activating factor), and cytokines (interleukins and other bioresponse modifiers) which are responsible for the inflammatory response. Cytokines are low molecular weight extracellular polypeptides and glycoproteins that regulate homeostasis and signal-dependent functioning of immune cells. Cytokines are key players of inflammation and include interleukins (ILs), chemokines, interferons (IFNs), tumor necrosis factor (TNF), colony-stimulating factor (CSF), and growth factors (GFs). A wide array of inflammatory signaling responses is involved and cytokines can be broadly classified as pro-inflammatory or anti-inflammatory mediators.
4.2.1. Role of Nrf2 in the Regulation of Cytokines
LPS stimulation induces exaggerated massive expression of inflammatory mediators including cytokines, chemokines, and adhesion molecules in Nrf2 knockout mice [76]. For many years, it was believed that macrophages polarize into a pro-inflammatory ‘M1′ phenotype, promoting the secretion of pro-inflammatory mediators such as TNF, IL-1 and IL-6 and small molecule mediators of inflammation such as ROS, RNS and prostanoids but conversely inducing a simultaneous decrease in anti-inflammatory cytokines such as transforming growth factor (TGF)-α, IL-4, IL-13, and IL-10 [141]. The activation of Nrf2 signaling represents an interesting alternative method to direct resolution of inflammation. It was reported that activation of Nrf2 has the potential to inhibit LPS-induced up-regulation of pro-inflammatory cytokines including IL-6 and IL-1β, through the ROS-independent inhibition [142]. This connection was further corroborated by the observation that Nrf2 negatively regulates the NF-κB-mediated transcription of pro-inflammatory cytokine genes [142]. The binding sites of Nrf2 near the IL-6 and IL-1β promoters coincides with the common binding regions of pro-inflammatory transcription factors, such as p65, C/EBPβ and c-Jun, suggesting that the recruitment of Nrf2 in this region might involve the interaction with other factors. This type of association was firstly suggested by studies reporting that p65 is responsible for the regulation of Nrf2 signaling [142]. Other co-activators include CBP, and Mediator complex MED16 and MED24, which were also reportedly found to be associated with Nrf2 in LPS-stimulated cells [143,144]. Also, histone-modifying enzymes, such as CBP/p300, were identified as specific cofactors recruited to the promoter region upon DNA-binding of a signal-specific transcription factor, an event followed by the recruitment of Mediator complex and the subsequent expression of Nrf2 target genes [145]. It is plausible that the recruitment of Nrf2 in regulatory regions of pro-inflammatory cytokine genes might abrogate their transcription by interfering with their transcription factors. This hypothesis is supported by evidence showing that the Nrf2 inducer Tecfidera, inhibited the IL-6 and IL-1 gene expression in experimental models of multiple sclerosis and other autoimmune diseases [146,147,148,149]. A significant reduction in Th1 and Th17 cytokines including IL-6 was observed upon Nrf2 activation induced by derivatives of the triterpene oleanolic acid, namely CDDO-trifluoroethyl-amide (CDDO-TFEA) mediating the suppression of IL-6 gene expression [150].
A study in Nrf2 knock-down peritoneal neutrophils also indicated that LPS stimulation was followed by a significant elevation in cytokines (TNF-α, IL-6) and chemokines such as monocyte chemotactic protein-1 (MCP-1) and macrophage inflammatory protein 2 (Mip2) as well as increased ROS levels [151]. In addition, it was also observed that the expression of GCLC, GCLM, and NQO1 was significantly up-regulated in wild-type neutrophils as compared with Nrf2 deficient cells [151]. However, pretreatment of peritoneal neutrophils with the Nrf2 activator CDDO, induced Nrf2-dependent antioxidant genes and attenuated changes in proinflammatory cytokines and chemokines in the lungs [151]. Taken together, these results suggest that oxidative regulation by Nrf2 is responsible for LPS-TLR4 signaling and thereby is crucial for the innate immune response in neutrophils and macrophages. Similarly, Nrf2-dependent HO-1 expression was found to negatively regulate the NF-ĸB and MCP-1 secretion levels in human umbilical vein endothelial cells (HUVECs) upon TNF-α treatment [152].
Some interesting studies conducted on myeloid-derived suppressor cells (MDSCs) demonstrated that the Nrf2 antitumor activity involves the suppression of ROS levels and tumor metastasis caused by impaired IL-6 secretion in MDSCs [153,154]. The ROS and RNS generation was reported to suppress the activation and the proliferation of CD8+ T-cells by inducing modifications in the T-cell receptor-CD8 complex upon the surface of CD8+ T-cells due to the production of peroxynitrite in MDSCs [155]. These changes disrupted the interaction between major histocompatibility complex class I molecules and the antigen-presenting cells [155], suggesting that the immunological response was implicated in the anti-metastatic activity of Nrf2.
It was also reported that Nrf2 activation by Keap1 knockout protects from obesity-induced diabetes by improving both insulin secretion and insulin resistance, thus resulting in the prevention of hyperglycemia [156]. Another report demonstrated that the improvement of insulin secretion and protection in type-2 diabetes was associated with the suppression of IL-1 and IL-1 receptor [157].
4.2.2. Role of Nrf2 in the Regulation of Cell Adhesion Molecules
In the pathological inflammatory process, IL-1 and TNF-α, reportedly up-regulate the expression of cell adhesion molecules (CAMs), such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) on the surface of endothelial cells, macrophages, and lymphocytes. All these molecules are responsible for leukocyte recruitment and cytokine production resulting in the tissue inflammation and oxidative stress [158,159]. Based on current knowledge, it is conceivable that Nrf2 might serve as a regulatory factor affecting the expression of cell adhesion molecules (CAMs). Indeed, Keap1 knockdown in HUVECs was shown to significantly attenuate the TNF-α induced increase in the expression of ICAM-1 and VCAM-1, suggesting the crucial role of Nrf2 in the vascular inflammatory responses [160]. Consistently, in response to LPS- administration in mice, Keap1 siRNA resulted in a significant and Nrf2-dependent down-regulation in the levels of TNF-α, iNOS, and several adhesion molecules [160]. On the other hand, Keap1 knockdown enhanced the expression of HO-1, GCL, and peroxiredoxin-1 (Prx1) in macrophage [160]. Knockdown of Nrf2 was also shown to stimulate NFκB activation, TNF-α, IL-1β, and IL-6 secretion and to promote the over-expression of ICAM-1 in the brain following traumatic brain injury [161].
Similarly, studies on vascular inflammation and atherosclerosis models revealed that activated Nrf2 can prevent the pro-inflammatory state of vascular endothelial cells by suppressing the p38–VCAM-1 signaling [162]. Here, Nrf2 inactivated p38 by inhibiting MKK3/6 signaling pathway and by inducing the activity of a negative regulator MKP-1 [162]. Moreover, MKP-1 is preferentially expressed by endothelial cells in aorta region where it inhibits p38–VCAM-1 signaling [163]. In this regard, some studies demonstrated the importance of the antioxidant enzyme HO-1, suggesting that the anti-inflammatory action of Nrf2 might in part depend on the modulation of the redox homeostasis. For instance, the plant-derived antioxidant 3-hydroxyanthranilic acid (HA), was found to induce HO-1 expression and to inhibit both VCAM-1 expression and NF-kB activation by promoting Nrf2 translocation to suppress inflammation in atherosclerosis [152]. Consistently, the genetic over-expression of HO-1 was found to inhibit TNF-α-mediated VCAM-1 expression in HUVECs [39]. In agreement with this hypothesis, the anti-inflammatory activity of several natural antioxidants such as lycopene, arachidin-1 and others, were shown to involve the inhibition of NF-κB transcription and ICAM-1 and VCAM-1 expression via Nrf2 dependent signaling pathway [41,164,165]. The Nrf2-dependent modulation of adhesion molecules plays also an important role in mediating cancer cells invasion and adaptation to persistent inflammatory stress during tumorigenesis. Indeed, it was reported that in the context of the inflammation associated with carcinogenesis, Nrf2 activation was able to abrogate the expression of E-cadherin, whereas in contrast to slightly increase the expression of L1CAM in pancreatic ductal adenocarcinoma cell lines [166]. Moreover, this report also identified a ARE region located in the promoter region of the E-cadherin gene which was involved in the Nrf2 dependent regulation of E-cadherin expression [166].
These findings indicate that in response to inflammatory stimuli, up-regulation of Nrf2 signaling inhibits the activation of NF-ĸB and the secretion of pro-inflammatory cytokines and chemokines, attenuating also the expressions of CAMs.
In addition to cytokines, chemokines, and CAMs, inflammatory mediators also include enzymes, which play a crucial role in tissue remodeling and inflammatory response. Activated leukocytes and mast cells express a variety of proteolytic enzymes, particularly serine proteases that ultimately modulate an inflammatory response [167].
4.2.3. Role of Nrf2 in the Regulation of Protease/Antiprotease Balance
Since the imbalance between protease and antiprotease levels and oxidative stress are also thought to be an important aspect of inflammation, Nrf2 is believed to play a regulatory mechanism in this aspect as well. In this respect, it was reported that the susceptibility of Nrf2 knock-out mice to emphysema was caused by a reduced antiprotease activity (Ishii et al., 2005). Mechanistically, Nrf2 knock-out mice showed a significant elevation in neutrophil elastase activity, suggesting protease/antiprotease imbalance (Ishii et al., 2005). The same group also revealed that secretory leukoprotease inhibitor (SLPI) levels were significantly suppressed in Nrf2 knock-out mice, whereas the SLPI gene was highly inducible in the lungs of wild-type mice after porcine pancreatic elastase induced emphysema [168]. By controlling the levels of this crucial regulator, Nrf2 is therefore expected to modulate both antiprotease and anti-inflammatory events in lung inflammation. For example, an earlier study showed that SLPI was markedly expressed at the site of inflammation in response to several inflammatory stimuli such as LPS [169] and proinflammatory cytokines [170]. Quite recent data suggest that SLPI might be a direct target of Nrf2 since its promoter was found to contain ARE regions which were responsible for the overexpression of SLPI by Nrf2 agonists in airway epithelial cells in an Nrf2-dependent manner [171,172].
Another type of inflammatory mediators implicated also in the malignant progression is represented by the matrix metalloproteinases (MMPs), zinc-dependent proteolytic enzymes responsible for the degradation of various components of extracellular matrix components. The activities of MMPs are regulated at multiple levels, including Nrf2 signaling. A series of experiments in Nrf2 knockout mice have demonstrated its protective role in UV-induced inflammation and the regulation of MMPs activities. For instance, MMP-9 activity and expression of MIP-2, a key mediator of neutrophil recruitment as well as Pro-MMP-9 expression were significantly elevated in Nrf2 knockout mice after UVB irradiation, suggesting that the photo-protective effect of Nrf2 was mediated by the suppression of MMPs expression [173]. Furthermore, Nrf2 could influence MMP-1 expression through the MAPK/AP-1 signaling cascades after UVA-induced stimulation in human keratinocytes (HaCaT cells) [174]. The study revealed that Nrf2-activating compounds could reverse the UVA-stimulated increase in phosphorylation of ERK, JNK, p38, c-Jun, and c-Fos [174]. Another study suggested that Nrf2 deficiency could induce the activation of MAPKs, such as JNK, ERK, and p38, in addition to c-Fos in osteoclast differentiation elicited by pro-inflammatory stimuli, suggesting that Nrf2 activation might be beneficial to treat inflammatory bone diseases [175].
Nrf2/HO-1 signaling inhibits the expression of MMP-9 and iNOS in LPS-stimulated macrophages [176] and IL-8, ICAM-1, COX-2 and MMP-7 expression in TNF-α-stimulated human intestinal epithelial HT-29 cells as well as in colonic mucosal tissue [177]. In addition, Nrf2 deficient mice subdued to spinal cord injury show significant elevation in NF-κB activation, TNF-α production, and expression of MMP-9 as compared with wild-type controls [178]. Similarly, in osteoarthritis model, Nrf2-knockout induces proinflammatory cytokines (TNF-α, IL-6, and IL-1β), MMP1, MMP3, and MMP13 expressions in chondrosarcoma cells [179]. In the treatment of diabetic skin ulcers, knockdown of Nrf2 shows delayed wound healing, which is attributed to oxidative DNA damage, reduced TGF-β1 levels, high MMP9 expression and increased apoptosis [180].
Nrf2 was also shown to modulate the expression of CD36 in macrophages. CD36 is a class B transmembrane scavenger receptor expressed by macrophages, microvascular endothelial cells, platelets, and epithelial cells [181]. CD36 is a major endocytic receptor for oxidized lipoproteins and is responsible for the uptake of oxidized LDLs (oxLDLs) by macrophages [181]. In macrophages, CD36 is reportedly up-regulated by oxLDL [182] and oxLDL induce the activation of Nrf2 [183]. The lack of induction of CD36 in Nrf2 knockout macrophages was found to be associated with a reduced accumulation of cholesterol, suggesting a role for the Nrf2 signaling pathway in modulating oxLDL uptake via CD36 [183]. Some other studies also indicate elevation in the expression of CD36 via Nrf2 activation in atherogenesis [184,185,186]. In an intracerebral hemorrhage, the Nrf2 activator sulforaphane induced CD36 expression in the affected brain and improved hematoma clearance in wild-type but not in Nrf2 knockout mice [187].
The inflammatory response also requires a systematic interaction of inflammatory enzymes known as cyclooxygenases (COX) which catalyze the formation of prostaglandins, thromboxane, and levuloglandins [188]. Among all the COX enzymes, COX-2 is mainly involved in acute inflammation [189]. Initially, COX-2 is mainly expressed by macrophages and is up-regulated in response to growth factors or inflammatory stimuli. A potential role of Nrf2 in repressing COX signaling is shown by the fact that the expression of COX-2, iNOS, and proinflammatory cytokines is significantly enhanced in Nrf2 knockout mice compared to their wild-type counterpart [190,191]. Similarly, a significant increase in oxidative stress and expression of COX-2, iNOS, IL-6, and TNF-α was observed after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine stimulation in Nrf2-knockout mice [121]. Likewise, some studies also show that the anti-inflammatory role of Nrf2 was reportedly mediated by COX2/15d-PGJ2 signaling pathway in experimental pleurisy and acute lung injury models [192,193].
Cyclopentenone PGs (cyPGs) such as 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) were also found to modulate Nrf2 signaling and to induce GST in peritoneal macrophages. Here, it was postulated that the presence of highly reactive α,β-unsaturated carbonyl group might confer to cyPGs the ability to form Michael adducts with nucleophilic molecules and to induce covalent modifications of target proteins, a mechanism that might be responsible for the activation of Nrf2 [192]. 15d-PGJ2 was also reported to induce the expression of HO-1 in a Nrf2 dependent manner [194]. Since the redox-sensitive cysteines are highly susceptible to adduct formation with electrophiles, CyPGs including 15d-PGJ2 can directly bind to the cysteines within the Keap1 IVR region, resulting in the activation of Nrf2/ARE signaling pathway [195,196,197]. Moreover, the anti-inflammatory activity of 15d-PGJ2 against acute lung injury was shown to be mediated by the induction of the Nrf2 pathway and this protective effect was abolished in Nrf2 knockout mice [192]. Similarly, pretreatment with Nrf2 activators such as sulforaphane, can exert anti-inflammatory effects by suppressing the expression of proinflammatory cytokines, COX-2, and iNOS in peritoneal macrophages, while this effect was abolished in Nrf2 knockout mice [198]. In human aortic endothelial cells (HAECs), 15d-PGJ2 activates Nrf2 and induces Nrf2 target genes expression [196]. COX-2 specific inhibitors including NS-398 were found to attenuate Nrf2 nuclear accumulation in the acute phase of laminar shear stress [196]. Therefore, crosstalk between the Nrf2 and signaling of inflammatory mediators needs further investigation.
5. Incongruity in the Role of Nrf2 in Inflammasome Signaling
The pyrin domain containing 3 (NLRP3) inflammasome consists of an amino-terminal pyrin domain (PYD), a central NACHT domain and a carboxy-terminal leucine-rich repeat domain (LRR domain) [199]. NLRP3 acts as a sensor that detects a broad range of pathological signals including pathogen-associated molecular patterns (PAMPs), damage-associated molecular pattern molecules (DAMPs), cytosolic or mitochondrial ROS, microbial motifs, and many environmental signals [199]. Activated NLRP3 inflammasome, in turn, activates caspase 1, which results in the cleavage of pro- IL-1β and pro- IL-18 [200]. Caspases 4, 5 and 11 are also activated and subsequently cleave the substrate gasdermin D (GSDMD), which in turn inserts into the membrane thereby forming pores that ultimately trigger pyroptosis [201].
Multiple studies investigated potential mechanisms underlying the Nrf2-dependent regulation of NLRP3 inflammasome. Since Nrf2 is involved in the regulation of oxidative stress and antioxidant genes expression, it is believed to inhibit the activation of NLRP3 by suppressing ROS production [202]. Additionally, Nrf2 down-regulates the expression of genes involved in inflammasome assembly such as NLRP3, caspase 1, IL-1β, and IL-18, thereby inhibiting NLRP3 inflammasome activity [203]. Importantly, the known Nrf2 activator tert-butylhydroquinone (tBHQ), was found to induce the expression of NQO1 and to inhibit NLRP3 priming in Nrf2-ARE dependent manner [203].
These data suggest Nrf2 as a novel therapeutic target to inhibit the assembly of the NLRP3 inflammasome complex. In this context, several herbal extracts, and plant-derived compounds such as epigallocatechin-3-gallate and citral (3,7-dimethyl-2,6-octadienal) were found to suppress NLRP3 inflammasome activation in various animal models by engaging the Nrf2 signaling pathway [204,205,206]. For example, a flavonoid compound, isoliquiritigenin, was shown to inhibit ROS and activate Nrf2/ARE signaling which in turn suppressed the NLRP3 and NF-κB pathways [207]. Another natural polyphenol, mangiferin, inhibited several steps of the LPS/D-GalN induced NLRP3 inflammasome assembly complex including caspase-1, IL-1β, and TNF-α by promoting Nrf2 dependent HO-1 expression [208]. A very recent investigation showed that melatonin similarly was able to trigger Nrf2 signaling which in turn inhibited NLRP3 expression, ASC (apoptosis-associated speck-like protein) formation, caspase-1 cleavage, and Il-1β maturation and secretion [209]. In contrast; however, the activation of Nrf2 signaling by sulforaphane was shown to inhibit Nlrp3 inflammasome in bone marrow-derived macrophages (BMDMs) in Nrf2 independent manner [210]. While the negative modulation of inflammasome by sulforaphane was also confirmed in Alzheimer’s disease, the underlying mechanism appeared to be slightly different since this compound prevented Aβ1–42-induced caspase-1-dependent inflammasome activation, by suppressing STAT-1 phosphorylation, suggesting that multiple mechanisms might be involved in this pathway [211]. Similar results were also obtained in other studies testing the effects of sulforaphane and xanthohumol which essentially confirmed the inhibition of IL-1β by Nrf2/NQO1/HO-1 signaling pathway and GCL expression [212,213].
Importantly, there is contradictory evidence showing that Nrf2 activation is essential for the induction of NLRP3 and AIM2 inflammasome in LPS-primed BMDMs following different stimuli [214]. NLRP3 activators, such as silica, alum, cholesterol, and monosodium urate (MSU) crystals, failed to activate IL-1β secretion and the assembly of cytosolic ASC speck in Nrf2-deficient macrophages, suggesting that Nrf2 is essential for NLRP3 assembly under certain circumstances [214,215,216]. In addition to this, a role for Nrf2 mediated phagocytosis was also reported to play a role in the activation of NLRP3 and IL-β secretion in studies showing impaired phagocytosis in macrophages and high susceptibility to infections in Nrf2 knockout mice [217]. Consistent with this, another study reported the activation of NLRP3 inflammasome by silica crystals and aluminum salts via Nrf2 mediated phagocytosis [218].
However, it is still controversial whether activation of Nrf2 can lead to the prevention or the induction of aggressive inflammation in response to cholesterol or MSU treated macrophages. These conflicting results suggest the need to focus on the role of unknown functions of Nrf2 signaling pathway in hyperlipidemia, atherosclerosis, gout, and other inflammation-related diseases, which could enable the discovery of new therapeutics based on this pathway.
6. Nrf2 Dependent Anti-Inflammatory Drugs
Nonsteroidal anti-inflammatory drugs (NSAIDs) including traditional non-selective NSAIDS (nsNSAIDs) and cyclo-oxygenase-2 selective NSAIDs (COXIBs), are one of the most common medication for inflammation [219]. The primary activity of NSAIDs are enacted by blocking prostaglandins (PGs) synthesis through the cyclooxygenase enzymes (COX-1 and COX-2) inhibition. Aspirin is the most commonly used NSAID with antioxidant activity and reportedly activate Nrf2-ARE pathway and HO-1 expression in primary human melanocytes [220] as well as in neuronal apoptosis [221]. Similarly, celecoxib up-regulates heme oxygenase-1 (HO-1) expression in macrophages and vascular smooth muscle cells via redox signaling [222]. Treatment of celecoxib demonstrated increases in H-ferritin and TrxR1 mRNA levels respectively in Nrf2 dependent mechanism [223]. A widely used NSAID, diclofenac also activates Nrf2 expression and downstream related genes [224]. In choroidal neovascularization, indomethacin or bromfenac induces translocation of Nrf2 into the nucleus and up-regulates HO-1 expression [225]. On the other hand, pantoprazole, a proton pump inhibitor (PPIs) protected from NSAIDs-induced injury by inducing activation of Nrf2 through inactivation of Keap1 and by increasing expression of HO-1 in human gastric epithelial and endothelial cells and in animal model of gastric injury [226].
In vitro and in vivo studies on NO-NSAID (nitric oxide-donating NSAID) hybrid drugs such as NCX-4016 (mNO-ASA, nitric oxide-donating aspirin) and the isomeric NCX-4040, suggest significant potential for activation of Nrf2 through nitrosylation of Keap1 [227,228]. Moreover, cellular bioactivation of pNO-ASA yields a quinone methide, therefore, an alternative mechanism of Nrf2 activation by mNO-ASA is via an electrophilic quinone [229]. Analogues of NCX-4016 that are activated to quinone methide and without NO group, also showed covalent modification of Keap1, and induced Nrf2 translocation to the nucleus [230]. Accumulating evidence also suggests that treatment with small molecules such as 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid- derivatives and sulforaphane induces Nrf2 activation and subsequently reduces significantly Th1 and Th17 cytokines including IL-6 by inhibiting recruitment of RNA polymerase II [231]. Similarly, pharmacological activation of mycophenolate mofetil, prevented the proinflammatory cytokines overexpression in a Nrf2 dependent mechanism [232]. In addition, short peptides such as casein glycomacropeptide hydrolysate were shown to have anti-inflammatory and antioxidant activity by increasing the Nrf2 nuclear translocation and HO-1 expression in RAW 264.7 macrophages and HepG2 cells [233,234]. Nrf2 induction by triazole derivatives inhibits pro- inflammatory cytokines release in different models of Huntington disease including primary mouse microglia, astrocytes, and Drosophila model and in cultured monocytes from human patients with huntington disease [235,236].
Several cell-derived metabolites such as itaconate and fumarate demonstrated anti-inflammatory responses in LPS- stimulated macrophages [237]. A cell-permeable derivative of itaconate, 4-octyl-itaconate (4-OI) demonstrated in vivo anti-inflammatory activity by induction of Nrf2 [237]. Here, the derivative of fumarate, dimethyl fumarate (DMF), is the only drug approved by US Food and Drug Administration and marketed by Biogen, as an anti-inflammatory therapeutic agent in multiple sclerosis with the ability to inhibit inflammation via Nrf2 antioxidant pathway [238,239]. In vivo studies in rodents had reported that DMF metabolite, monomethyl fumarate, activates Nrf2 by adduct formation at C151 in Keap1 in neuroinflammation [238]. Other commercial prodrugs of monomethyl fumarate such as diroximel fumarate is in a phase III clinical trial for multiple sclerosis and tepilamide fumarate, is in a phase II clinical trial for plaque psoriasis [240]. V ClinBio developed conjugate of monomethyl fumarate and eicosapentaenoic acid for simultaneous modulation of Nrf2 and NF-κB in cell lines and animal models of multiple sclerosis and psoriasis. Preclinical studies by Colby Pharmaceuticals developed a di- substituted hydroxylamine compound (OT551) which inhibits inflammation and oxidative stress by targeting Keap1 (OMEGA/NCT00485394). An ophthalmic solution of this compound protects from inflammation in retinal pigment epithelium and photoreceptors (OMEGA/NCT00485394).
7. Polyphenols and Nrf2 Signaling Can Modulate Inflammation
So far, we discussed different aspects of Nrf2 signaling pathway in inflammation, therefore, it is also worthwhile to discuss natural antioxidants which could modulate Nrf2-dependent treatment of inflammatory diseases [241]. In this context, dietary polyphenols are gaining interest and numerous studies supported their potential in animal and cell models. Polyphenols are secondary metabolites found in fruit and vegetables and are present either as glycosides esters or as free aglycones [241]. Several lines of evidence revealed that polyphenols could modulate oxidative stress and inflammation by inducing Nrf2 [242,243]. Curcumin, a polyphenol-derived compound, exhibits anti-inflammatory activity by Nrf2 activation and up-regulation of HO-1 expression [244]. A recent study demonstrated that curcumin inhibits the expression of both the inflammation mediators and of the matrix-degrading proteinases in inflammatory chondrocytes via ROS/Nrf2/HO-1-SOD2-NQO-1-GCLC signaling pathway [245]. In addition, bisdemethoxycurcumin, curcumin analog, induces activation of HO-1 expression through Ca2+/calmodulin-dependent protein kinase II–ERK1/2-Nrf2 cascade in LPS-stimulated macrophages [246]. Furthermore, demethoxy curcuminoids regulate Nrf2 mediated HO-1 expression via PI3K/Akt signaling pathway in β-cells [247]. Eriodictyol, a flavanone compound, exhibit anti-inflammatory and antioxidant activity by regulating Nrf2, inhibiting NF-ĸB, and inhibiting the expression of cytokines in macrophages [248,249]. Similarly, numerous phytochemicals such as resveratrol, lycopene, and luteolin exert prominent anti-inflammatory and antioxidant properties through the modulation of Nrf2 signaling pathway [250,251,252].
Epigallocatechin gallate (EGCG), another potent catechin antioxidant, ameliorates oxidative stress and inflammation by inducing both NF-κB and Nrf2 nuclear translocation and also promotes HO-1 expression in renal injury [253]. A similar trend of activation of Nrf2, ERK and p38 MAPK signaling pathways by EGCG was also observed against inflammation in different disease models such as oxalate-induced epithelial mesenchymal transition [254], obstructive nephropathy [253], fluoride induced lung inflammation [255] and diabetic nephropathy [256].
Flavonoids such as quercetin, naringenin and hesperidin-methyl-chalcone are well known for their anti-inflammatory properties. Hesperidin-methyl-chalcone inhibits oxidative stress and the release of inflammatory cytokines by inducing Nrf2 activation and HO-1 mRNA expression in gout induced inflammation in mice [257]. Similarly, naringenin promotes Nrf2/HO-1 signaling axis via PI3K/Akt signaling pathway in D-galactose-induced brain aging in mice [258]. Quercetin also demonstrated activation of Nrf2 and phase 2 enzymes HO-1, GPx1 and glutathione reductase (GR) in chronic arthritis [259,260]. Quercetin promotes Nrf2 activation by both Keap1-dependent and Keap1-independent mechanisms. Keap1-dependent mechanism involves the interaction of H-benzene and H-bond of quercetin to Keap-1 Arg415 and Tyr527, and Gly364, respectively. However, the Keap1-independent mechanism involves activation of JNK MAP kinase signaling pathway. Rosmarinic acid, a polyphenol-containing catechol, induces Nrf2 activation and attenuates chronic constriction injury (CCI)-induced neuropathic inflammation [261]. Rosmarinic acid induces Nrf2 activation through Akt/GSK-3β/Fyn pathway in β-amyloid-induced oxidative stress [262].
Kaurenoic acid, a diterpene, attenuated LPS-induced acute lung injury, neutrophil recruitment, and inflammatory cytokine gene expression by inducing Nrf2 activation and up-regulates the NQO-1, HO-1 and GCLC expressions [263]. Glycyrrhizin, a pentacyclic triterpenoid induces p38/Nrf2-dependent activation of HO-1 in LPS-induced Raw 264.7 cells [264].
8. Future Prospects
A correct approach for future research in the evaluation of the pharmacodynamic action of NRF2 inducers should assess a wide range of genetic and biochemical biomarkers (e.g., induction of the NQO1 transcript; epigenetic regulators of histone deacetylase, markers related to oxidative stress; pro and anti-inflammatory cytokines, NF-κB signaling molecules) in a dose-response manner by using validated and standardized methodologies. Additionally, in the preclinical studies, new disease directed animal models that best recapitulate human diseases concerning Nrf2-related pathogenic mechanisms should be developed to facilitate screening of new chemical entities as Nrf2 modulators. It is well known that non-communicable diseases are characterized by multiple pathways and complexity. The molecular mechanisms and histopathological pathways that lead to disease are different, but in many cases overlap. The research on a common molecular pathway such as the altered Nrf2 activity would help to facilitate the screening of new chemical entities as Nrf2 modulators or to consider re-proposing existing drugs. An area of potential interest for developing a candidate drug in the Nrf2 pathway would be the research on the indirect hallmarks of Nrf2 activation in diseased tissues that would represent a more robust and reliable readout of dosage regimen and drug distribution. More detailed studies are necessary to better characterize potential safety issues and to understand whether sustained Nrf2 induction could lead to the development of other diseases such as cancer. Furthermore, the functional characterization of the 27 cysteines retained by Keap1 and in particular, the identification of most reactive cysteines may also shed some light on the compounds which may modify Keap1 making it incapable of interacting with Nrf2 at both high as well as low-affinity sites. A major pitfall in the drugs acting on C151 is that they may interact with redox reactive cysteines in other proteins as well. Therefore, hierarchical modeling of target cysteines could have a potential advantage. Also, the studies of in vitro assays could identify off-target effects. Another concern which needs to be addressed is the inability of Nrf2 activators to cross the blood-brain barrier for therapeutic use in neurological disorders. Additionally, the therapeutic use of many Nrf2 inhibitors that have led to promising results in pre-clinical models still needs to be conclusively proven in rigorous clinical trials.
9. Conclusions
Nrf2-Keap1 signaling pathway is the hallmark of redox signaling and controlled inflammation and plays an important role in cell metabolic flexibility. Here, we reviewed ongoing scientific literature about regulation of Nrf2 signaling pathway in different aspects of inflammation such as cytokines, chemokine releasing factors, MMPs, and other inflammatory mediators affecting the NF-kB and MAPK networks to control inflammation. However, there are still some mechanisms such as interactions between Nrf2 and JAK/STAT signaling that needs to be investigated. Although many pharmaceutical companies are currently targeting Keap1, the prime regulator of Nrf2, it is still challenging to enhance the targeting of these compounds and their activity against these multifactorial complex diseases. Natural Nrf2 activators derived from plant sources as well as synthetic anti-inflammatory drugs require further experimental validation. Research for efficient therapeutic agents promoting Nrf2 activation came up with some new drugs which have entered clinical trials and will undoubtedly provide advancement in the management of inflammation in the near future. However, efforts are still ongoing to find new small molecule Nrf2 inducers with the advantages of oral administration, high target specificity, safe, and high bioavailability, therefore, present review would enable refinement in our understanding of Nrf2 signaling pathway interplay with expressions of associated target genes and support the hypothesis that Nrf2 inducers have the strong potential as anti-inflammatory therapeutic agents.
Funding
This research received no external funding.
Acknowledgments
The authors are thankful to the European Molecular Biology Organization for providing fellowship to S.S.
Conflicts of Interest
The author declares no conflict of interest.
References
- Soehnlein, O.; Lindbon, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [PubMed]
- Janeway, C.A.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology: The Immune System in Health and Disease; Garland Science: New York, NY, USA, 2005. [Google Scholar]
- Yang, D.; Han, Z.; Oppenheim, J.J. Alarmins and immunity. Immunol. Rev. 2017, 280, 41–56. [Google Scholar] [PubMed]
- Noah, T.A.; Zachary, M.W.; Randy, J.N. Inflammation: Mechanisms, costs, and natural variation. Annu. Rev. Ecol. Evol. Syst. 2012, 43, 385–406. [Google Scholar]
- Kaulmann, A.; Bohn, T. Carotenoids, inflammation, and oxidative stress–implications of cellular signaling pathways and relation to chronic disease prevention. Nutr. Res. 2014, 34, 907–929. [Google Scholar]
- Kaplan, M.H. STAT signaling in inflammation. JAKSTAT 2013, 2, e24198. [Google Scholar]
- Krajka-Kuźniak, V.; Paluszczak, J.; Baer-Dubowska, W. The Nrf2-ARE signaling pathway: An update on its regulation and possible role in cancer prevention and treatment. Pharmacol. Rep. 2017, 69, 393–402. [Google Scholar]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell Biol. 2009, 29, 2658–2672. [Google Scholar]
- Theodore, M.; Kawai, Y.; Yang, J.; Kleshchenko, Y.; Reddy, S.P.; Villalta, F.; Arinze, I.J. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J. Biol. Chem. 2008, 283, 8984–8994. [Google Scholar]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell Biol. 2005, 25, 10895–10906. [Google Scholar]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobon-Velasco, J.C.; Devijver, H.; Garcia-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol. Cell Biol. 2012, 32, 3486–3499. [Google Scholar] [PubMed] [Green Version]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [PubMed] [Green Version]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [PubMed] [Green Version]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A. The Keap1-Nrf2pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell Biol. 2005, 25, 162–171. [Google Scholar]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar]
- Kansanen, E.; Kivel, A.M.; Levonen, A.L. Regulation of Nrf2-dependent gene expression by 15-deoxy-Delta 12,14-prostaglandin J2. Free Radic. Biol. Med. 2009, 47, 1310–1317. [Google Scholar]
- Ogura, T.; Tong, K.I.; Mio, K.; Maruyama, Y.; Kurokawa, H.; Sato, C.; Yamamoto, M. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc. Natl. Acad. Sci. USA 2010, 107, 2842–2847. [Google Scholar]
- Zipper, L.M.; Mulcahy, R.T. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J. Biol. Chem. 2002, 277, 36544–36552. [Google Scholar]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cell. 2011, 16, 123–140. [Google Scholar]
- Um, H.C.; Jang, J.H.; Kim, D.H.; Lee, C.; Surh, Y.J. Nitric oxide activates Nrf2 through S-nitrosylation of Keap1 in PC12 cells. Nitric Oxide 2011, 25, 161–168. [Google Scholar]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-Sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar]
- Kansanen, E.; Jyrkkanen, H.K.; Levonen, A.L. Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radic. Biol. Med. 2012, 52, 973–982. [Google Scholar] [PubMed]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Gene Dev. 1999, 13, 76–86. [Google Scholar]
- Kim, J.H.; Yu, S.; Chen, J.D.; Kong, A.N. The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains. Oncogene 2013, 32, 514–527. [Google Scholar]
- Rachakonda, G.; Xiong, Y.; Sekhar, K.R.; Stamer, S.L.; Liebler, D.C.; Freeman, M.L. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem. Res. Toxicol. 2008, 21, 705–710. [Google Scholar]
- Chowdhry, S.; Zhang, M.; McMahon, Y.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}- TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [PubMed] [Green Version]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [PubMed] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Sutter, T.R.; Kensler, T.W. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1,2-dithiole-3-thione. Mol. Med. 2001, 7, 135–145. [Google Scholar] [PubMed]
- Vollrath, V.; Wielandt, A.M.; Iruretagoyena, M.; Chianale, J. Role of Nrf2 in the regulation of the Mrp2 (ABCC2) gene. Biochem. J. 2006, 395, 599–609. [Google Scholar] [PubMed] [Green Version]
- Gilmore, T. The Rel/NF-κB signal transduction pathway: Introduction. Oncogene 1999, 18, 6842–6844. [Google Scholar] [PubMed] [Green Version]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-kappa B system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [PubMed]
- Soares, M.P.; Seldon, M.P.; Gregoire, I.P.; Vassilevskaia, T.; Berberat, P.O.; Yu, J.; Tsui, T.Y.; Bach, F.H. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J. Immunol. 2004, 172, 3553–3563. [Google Scholar]
- Yerra, V.G.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397. [Google Scholar]
- Chen, L.G.; Zhang, Y.Q.; Wu, Z.Z.; Hsieh, C.W.; Chu, C.S.; Wung, B.S. Peanut arachidin-1 enhances Nrf2-mediated protective mechanisms against TNF-α-induced ICAM-1 expression and NF-κB activation in endothelial cells. Int. J. Mol. Med. 2018, 41, 541–547. [Google Scholar]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappab/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [PubMed] [Green Version]
- Brigelius-Flohé, R.; Flohé, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011, 15, 2335–2381. [Google Scholar]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and their concerted modulation in cancer pathogenesis and progression. Cancer 2010, 2, 483–497. [Google Scholar]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar]
- Chuang, H.C.; Chang, C.W.; Chang, G.D.; Yao, T.P.; Chen, H. Histone deacetylase 3 binds to and regulates the GCMa transcription factor. Nucl. Acids Res. 2006, 34, 1459. [Google Scholar]
- Hung, H.L.; Kim, A.Y.; Hong, W.; Rakowski, C.; Blobel, G.A. Stimulation of NF-E2 DNA binding by CREB-binding protein (CBP)-mediated acetylation. J. Biol. Chem. 2001, 276, 10715. [Google Scholar]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868. [Google Scholar]
- Go, Y.M.; Gipp, J.J.; Mulcahy, R.T.; Jones, D.P. H2O2-dependent activation of GCLC-ARE4 reporter occurs by mitogen-activated protein kinase pathways without oxidation of cellular glutathione or thioredoxin-1. J. Biol. Chem. 2004, 279, 5837. [Google Scholar]
- Carayol, N.; Chen, J.; Yang, F.; Jin, T.; Jin, L.; States, D.; Wang, C.Y. A dominant function of IKK/NF-kappaB signaling in global lipopolysaccharide-induced gene expression. J. Biol. Chem. 2006, 281, 31142. [Google Scholar]
- Hosoya, T.; Maruyama, A.; Kang, M.I.; Kawatani, Y.; Shibata, T.; Uchida, K.; Warabi, E.; Noguchi, N.; Itoh, K.; Yamamoto, M. Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J. Biol. Chem. 2005, 280, 27244–27250. [Google Scholar]
- Rushworth, S.A.; Chen, X.L.; Mackman, N.; Ogborne, R.M.; O’Connell, M.A. Lipopolysaccharide-induced heme oxygenase-1 expression in human monocytic cells is mediated via Nrf2 and protein kinase C. J. Immunol. 2005, 175, 4408. [Google Scholar] [PubMed] [Green Version]
- Ahn, K.S.; Aggarwal, B.B. Transcription Factor NF-{kappa}B: A sensor for smoke and stress signals. Ann. N. Y. Acad. Sci. 2005, 1056, 218. [Google Scholar] [PubMed]
- Anwar, A.A.; Li, F.Y.; Leake, D.S.; Ishii, T.; Mann, G.E.; Siow, R.C. Induction of heme oxygenase 1 by moderately oxidized low-density lipoproteins in human vascular smooth muscle cells: Role of mitogen-activated protein kinases and Nrf2. Free Radic. Biol. Med. 2005, 39, 227. [Google Scholar] [PubMed]
- Knorr-Wittmann, C.; Hengstermann, A.; Gebel, S.; Alam, J.; Muller, T. Characterization of Nrf2 activation and heme oxygenase-1 expression in NIH3T3 cells exposed to aqueous extracts of cigarette smoke. Free Radic. Biol. Med. 2005, 39, 1438. [Google Scholar] [PubMed]
- Lan, Q.; Mercurius, K.O.; Davies, P.F. Stimulation of transcription factors NF kappa B and AP1 in endothelial cells subjected to shear stress. Biochem. Biophys. Res. Commun. 1994, 201, 950. [Google Scholar] [PubMed]
- Arinze, I.J.; Kawai, Y. Transcriptional activation of the human Galphai2 gene promoter through nuclear factor-kappaB and antioxidant response elements. J. Biol. Chem. 2005, 280, 9786. [Google Scholar]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NFkappaB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar]
- Minelli, A.; Grottelli, S.; Mierla, A.; Pinnen, F.; Cacciatore, I.; Bellezza, I. Cyclo(His-Pro) exerts anti-inflammatory effects by modulating NF-κB and Nrf2 signalling. Int. J. Biochem. Cell Biol. 2012, 44, 525–535. [Google Scholar]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.S.; Yu, S.; Kong, A.N. Activation of Nrf2- antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar]
- Rushworth, S.A.; MacEwan, D.J.; O’Connell, M.A. Lipopolysaccharide-induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J. Immunol. 2008, 181, 6730–6737. [Google Scholar]
- Iskander, K.; Li, J.; Han, S.; Zheng, B.; Jaiswal, A.K. NQO1 and NQO2 regulation of humoral immunity and autoimmunity. J. Biol. Chem. 2006, 281, 30917–30924. [Google Scholar] [PubMed] [Green Version]
- Das, K.C. c-Jun NH2-terminal kinase-mediated redox-dependent degradation of IkappaB: Role of thioredoxin in NF-kappaB activation. J. Biol. Chem. 2001, 276, 4662–4670. [Google Scholar] [PubMed] [Green Version]
- Freemerman, A.J.; Gallegos, A.; Powis, G. Nuclear factor kappaB transactivation is increased but is not involved in the proliferative effects of thioredoxin overexpression in MCF-7 breast cancer cells. Cancer Res. 1999, 59, 4090–4094. [Google Scholar] [PubMed]
- Hayashi, T.; Ueno, Y.; Okamoto, T. Oxidoreductive regulation of nuclear factor kappa B. Involvement of a cellular reducing catalyst thioredoxin. J. Biol. Chem. 1993, 268, 11380–11388. [Google Scholar]
- Matthews, J.R.; Wakasugi, N.; Virelizier, J.L.; Yodoi, J.; Hay, R.T. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucl. Acids Res. 1992, 20, 3821–3830. [Google Scholar]
- Hirota, K.; Murata, M.; Sachi, Y.; Nakamura, H.; Takeuchi, J.; Mori, K.; Yodoi, J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus: A two-step mechanism of redox regulation of transcription factor NF-kappaB. J. Biol. Chem. 1999, 274, 27891–27897. [Google Scholar]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar]
- Dai, Y.; Zhang, H.; Zhang, J.; Yan, M. Isoquercetin attenuates oxidative stress and neuronal apoptosis after ischemia/reperfusion injury via Nrf2-mediated inhibition of the NOX4/ROS/NF-kappaB pathway. Chem. Biol. Interact. 2018, 284, 32–40. [Google Scholar]
- Yamauchi, K.; Nakano, Y.; Imai, T.; Takagi, T.; Tsuruma, K.; Shimazawa, M.; Iwama, T.; Hara, H. A novel nuclear factor erythroid 2-related factor 2 (Nrf2) activator RS9 attenuates brain injury after ischemia reperfusion in mice. Neuroscience 2016, 333, 302–310. [Google Scholar]
- Chen, X.L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1862–H1870. [Google Scholar]
- Chen, X.L.; Varner, S.E.; Rao, A.S.; Grey, J.Y.; Thomas, S.; Cook, C.K.; Wasserman, M.A.; Medford, R.M.; Jaiswal, A.K.; Kunsch, C. Laminar flow induction of antioxidant response element-mediated genes in endothelial cells: A novel anti-inflammatory mechanism. J. Biol. Chem. 2003, 278, 703–711. [Google Scholar]
- Kanninen, K.; Heikkinen, R.; Malm, T.; Rolova, T.; Kuhmonen, S.; Leinonen, H.; Yla-Herttuala, S.; Tanila, H.; Levonen, A.L.; Koistinaho, M.; et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16505–16510. [Google Scholar] [PubMed] [Green Version]
- Levonen, A.L.; Inkala, M.; Heikura, T.; Jauhiainen, S.; Jyrkkanen, H.K.; Kansanen, E.; Maatta, K.; Romppanen, E.; Turunen, P.; Rutanen, J.; et al. Nrf2 gene transfer induces antioxidant enzymes and suppresses smooth muscle cell growth in vitro and reduces oxidative stress in rabbit aorta in vivo. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 741–747. [Google Scholar] [PubMed]
- Heiss, E.; Herhaus, C.; Klimo, K.; Bartsch, H.; Gerhauser, C. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J. Biol. Chem. 2001, 276, 32008–32015. [Google Scholar]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [PubMed] [Green Version]
- Rangasamy, T.; Guo, J.; Mitzner, W.A.; Roman, J.; Singh, A.; Fryer, A.D.; Yamamoto, M.; Kensler, T.W.; Tuder, R.M.; Georas, S.N.; et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 2005, 202, 47–59. [Google Scholar]
- Jin, W.; Zhu, L.; Guan, Q.; Chen, G.; Wang, Q.F.; Yin, H.X.; Hang, C.H.; Shi, J.X.; Wang, H.D. Influence of Nrf2 genotype on pulmonary NF-kappaB activity and inflammatory response after traumatic brain injury. Ann. Clin. Lab. Sci. 2008, 38, 221–227. [Google Scholar] [PubMed]
- Cho, H.Y.; Imani, F.; Miller-DeGraff, L.; Walters, D.; Melendi, G.A.; Yamamoto, M.; Polack, F.P.; Kleeberger, S.R. Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. Am. J. Respir. Crit. Care Med. 2009, 179, 138–150. [Google Scholar]
- Rushworth, S.A.; Shah, S.; MacEwan, D.J. TNF mediates the sustained activation of Nrf2 in human monocytes. J. Immunol. 2011, 187, 702–707. [Google Scholar]
- Yang, H.; Magilnick, N.; Lee, C.; Kalmaz, D.; Ou, X.; Chan, J.Y.; Lu, S.C. Nrf1 and Nrf2 regulate rat glutamate-cysteine ligase catalytic subunit transcription indirectly via NF-kappaB and AP-1. Mol. Cell Biol. 2005, 25, 5933–5946. [Google Scholar]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [PubMed] [Green Version]
- Brewer, A.C.; Murray, T.V.A.; Arno, M.; Zhang, M.; Anilkumar, N.P.; Mann, G.E.; Shah, A.M. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic. Biol. Med. 2011, 51, 205–215. [Google Scholar] [PubMed] [Green Version]
- Strom, J.; Xu, B.; Tian, X.; Chen, Q.M. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 2016, 30, 66–80. [Google Scholar] [PubMed] [Green Version]
- Ross, D.; Siegel, D. Functions of NQO1 in cellular protection and CoQ10 metabolism and its potential role as a redox sensitive molecular switch. Free Physiol. 2017, 8, 595. [Google Scholar]
- Jaiswal, A.K. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic. Biol. Med. 2000, 29, 254–262. [Google Scholar]
- Iskander, K.; Barrios, R.J.; Jaiswal, A.K. Disruption of NAD(P)H:quinone oxidoreductase 1 gene in mice leads to radiation induced myeloproliferative disease. Cancer Res. 2008, 68, 7915–7922. [Google Scholar]
- Li, X.; Liu, Z.; Zhang, A.; Han, C.; Shen, A.; Jiang, L.; Boothman, D.A.; Qiao, J.; Wang, Y.; Huang, X.; et al. NQO1 targeting prodrug triggers innate sensing to overcome checkpoint blockade resistance. Nat. Commun. 2019, 10, 3251. [Google Scholar]
- Das, A.; Kole, L.; Wang, L.; Barrios, R.; Moorthy, B.; Jaiswal, A.K. BALT development and augmentation of hyperoxic lung injury in mice deficient in NQO1 and NQO2. Free Radic. Biol. Med. 2006, 40, 1843–1856. [Google Scholar]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Nat. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar]
- Kim, Y.J.; Ahn, J.Y.; Liang, P.; Ip, C.; Zhang, Y.; Park, Y.M. Human prx1 gene is a target of Nrf2 and is upregulated by hypoxia/reoxygenation: Implication to tumor biology. Cancer Res. 2007, 67, 546–554. [Google Scholar] [PubMed] [Green Version]
- He, X.; Ma, Q. Redox regulation by nuclear factor erythroid 2-related factor 2: Gatekeeping for the basal and diabetes-induced expression of thioredoxin-interacting protein. Mol. Pharmacol. 2012, 82, 887–897. [Google Scholar] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Ann. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Rad. Biol. Med. 2009, 46, 443–453. [Google Scholar] [PubMed] [Green Version]
- Rundlof, A.K.; Carlsten, M.; Arner, E.S. The core promoter of human thioredoxin reductase 1: Cloning, transcriptional activity, and Oct-1, Sp1, and Sp3 binding reveal a housekeeping-type promoter for the AUrich element-regulated gene. J. Biol. Chem. 2004, 276, 30542–30551. [Google Scholar]
- Bae, S.H.; Sung, S.H.; Cho, E.J.; Lee, S.K.; Lee, H.E.; Woo, H.A.; Yu, D.Y.; Kil, I.S.; Rhee, S.G. Concerted action of sulfiredoxin and peroxiredoxin I protects against alcohol-induced oxidative injury in mouse liver. Hepatology 2011, 53, 945–953. [Google Scholar]
- Moinova, H.R.; Mulcahy, R.T. An electrophile responsive element (EpRE) regulates betanaphthoflavone induction of the human gamma-glutamylcysteine synthetase regulatory subunit gene. Constitutive expression is mediated by an adjacent AP-1 site. J. Biol. Chem. 1998, 273, 14683–14689. [Google Scholar]
- Hartl, D.; Starosta, V.; Maier, K.; Beck-Speier, I.; Rebhan, C.; Becker, B.F.; Latzin, P.; Fischer, R.; Ratjen, F.; Huber, R.M.; et al. Inhaled glutathione decreases PGE2 and increases lymphocytes in cystic fibrosis lungs. Free Radic. Biol. Med. 2005, 39, 463–472. [Google Scholar]
- Manna, S.K.; Kuo, M.T.; Aggarwal, B.B. Overexpression of gamma-glutamylcysteine synthetase suppresses tumor necrosis factor-induced apoptosis and activation of nuclear transcription factor-kappa B and activator protein-1. Oncogene 1999, 18, 4371–4382. [Google Scholar]
- Zhang, F.; Wang, X.; Wang, W.; Li, N.; Li, J. Glutamine reduces TNF-alpha by enhancing glutathione synthesis in lipopolysaccharide-stimulated alveolar epithelial cells of rats. Inflammation 2008, 31, 344–350. [Google Scholar]
- Sun, S.; Zhang, H.; Xue, B.; Wu, Y.; Wang, J.; Yin, Z.; Luo, L. Protective effect of glutathione against lipopolysaccharide-induced inflammation and mortality in rats. Inflamm. Res. 2006, 55, 504–510. [Google Scholar] [PubMed]
- Peterson, J.D.; Herzenberg, L.A.; Vasquez, K.; Waltenbaugh, C. Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 3071–3076. [Google Scholar] [PubMed] [Green Version]
- Murata, Y.; Shimamura, T.; Hamuro, J. The polarization of T(h)1/T(h)2 balance is dependent on the intracellular thiol redox status of macrophages due to the distinctive cytokine production. Int. Immunol. 2002, 14, 201–212. [Google Scholar]
- Kim, Y.C.; Masutani, H.; Yamaguchi, Y.; Itoh, K.; Yamamoto, M.; Yodoi, J. Hemin-induced activation of the thioredoxin gene by Nrf2. A differential regulation of the antioxidant responsive element by a switch of its binding factors. J. Biol. Chem. 2001, 276, 18399–18406. [Google Scholar] [PubMed] [Green Version]
- Jiménez-Osorio, A.S.; González-Reyes, S.; García-Niño, W.R.; Moreno-Macías, H.; Rodríguez-Arellano, M.E.; Vargas-Alarcón, G.; Zúñiga, J.; Barquera, R.; Pedraza-Chaverri, J. Association of Nuclear Factor-Erythroid 2-Related Factor 2, Thioredoxin interacting protein, and heme oxygenase-1 gene polymorphisms with diabetes and obesity in Mexican patients. Oxid. Med. Cell. Longev. 2016, 2016, 7367641. [Google Scholar] [PubMed]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar]
- Sato, H.; Fujiwara, K.; Sagara, J.; Bannai, S. Induction of cystine transport activity in mouse peritoneal macrophages by bacterial lipopolysaccharide. Biochem. J. 1995, 310, 547–551. [Google Scholar]
- Manandhar, S.; Cho, J.M.; Kim, J.A.; Kensler, T.W.; Kwak, M.K. Induction of Nrf2-regulated genes by 3H-1, 2-dithiole-3-thione through the ERK signaling pathway in murine keratinocytes. Eur. J. Pharmacol. 2007, 577, 17–27. [Google Scholar]
- Primiano, T.; Kensler, T.W.; Kuppusamy, P.; Zweier, J.L.; Sutter, T.R. Induction of hepatic heme oxygenase-1 and ferritin in rats by cancer chemopreventive dithiolethiones. Carcinogenesis 1996, 17, 2291–2296. [Google Scholar]
- Kerins, M.J.; Ooi, A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [PubMed] [Green Version]
- Gu, J.; Cheng, Y.; Wu, H.; Kong, L.; Wang, S.; Xu, Z.; Zhang, Z.; Tan, Y.; Keller, B.B.; Zhou, H.; et al. Metallothionein is downstream of Nrf2 and partially mediates sulforaphane prevention of diabetic cardiomyopathy. Diabetes 2017, 66, 529–542. [Google Scholar] [PubMed] [Green Version]
- Reichard, J.F.; Motz, G.T.; Puga, A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nuc. Acids Res. 2007, 35, 7074–7086. [Google Scholar]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. Regulation of the Keap1–Nrf2 pathway by p62/SQSTM1. Curr. Opin. Toxicol. 2020, 1, 54–61. [Google Scholar]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar]
- Wunder, C.; Potter, R.F. The heme oxygenase system: Its role in liver inflammation. Curr. Drug Target. Cardiovasc. Haematol. Disord. 2003, 3, 199–208. [Google Scholar]
- Filardi, E.S.; Vega, M.A.; Mateos, P.S.; Corbi, A.L.; Kroger, A.P. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology 2010, 215, 788–795. [Google Scholar]
- Choi, K.M.; Kashyap, P.C.; Dutta, N.; Stoltz, G.J.; Ordog, T.; Donohue, T.S.; Bauer, A.J.; Linden, D.R.; Szurszewski, J.H.; Gibbons, S.J.; et al. CD206-positive M2 macrophages that express heme oxygenase-1 protect against diabetic gastroparesis in mice. Gastroenterology 2010, 138, 2399–2409. [Google Scholar]
- Nakamichi, I.; Habtezion, A.; Zhong, B.; Contag, C.H.; Butcher, E.C.; Omary, M.B. Hemin-activated macrophages home to the pancreas and protect from acute pancreatitis via heme oxygenase-1 induction. J. Clin. Investig. 2005, 115, 3007–3014. [Google Scholar]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar]
- Ho, F.M.; Kang, H.C.; Lee, S.T.; Chao, Y.; Chen, Y.C.; Huang, L.J.; Lin, W.-W. The anti-inflammatory actions of LCY-2-CHO, a carbazole analogue, in vascular smooth muscle cells. Biochem. Pharmacol. 2007, 74, 298–308. [Google Scholar] [PubMed]
- Luo, J.F.; Shen, X.Y.; Lio, C.K.; Dai, Y.; Cheng, C.S.; Liu, J.X.; Yao, Y.D.; Yu, Y.; Xie, Y.; Luo, P.; et al. Activation of Nrf2/HO-1 pathway by Nardochinoid C inhibits inflammation and oxidative stress in lipopolysaccharide-stimulated macrophages. Front. Pharmacol. 2018, 9, 911. [Google Scholar] [PubMed]
- Yet, S.F.; Perrella, M.A.; Layne, M.D.; Hsieh, C.M.; Maemura, K.; Kobzik, L.; Wiesel, P.; Christou, H.; Kourembanas, S.; Lee, M.E. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J. Clin. Investig. 1999, 103, R23–R29. [Google Scholar] [PubMed] [Green Version]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Pruthi, P.K.; Sawhney, S.; Piplani, T.; Wada, T.; Yachie, A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol. Oncol. 2011, 33, 74–78. [Google Scholar]
- Chi, X.; Yao, W.; Xia, H.; Jin, Y.; Li, X.; Cai, J.; Hei, Z. Elevation of HO-1 expression mitigates intestinal ischemia-reperfusion injury and restores tight junction function in a rat liver transplantation model. Oxid. Med. Cell Longev. 2015, 2015, 986075. [Google Scholar]
- Kim, W.; Kim, H.U.; Lee, H.N.; Kim, S.H.; Kim, C.; Cha, Y.N.; Joe, Y.; Chung, H.T.; Jang, J.; Kim, K.; et al. Taurine chloramine stimulates efferocytosis through upregulation of Nrf2-mediated heme oxygenase-1 expression in murine macrophages: Possible involvement of carbon monoxide. Antioxid. Redox Signal. 2015, 23, 163–177. [Google Scholar]
- Lea, S.; Plumb, J.; Metcalfe, H.; Spicer, D.; Woodman, P.; Fox, J.C.; Singh, D. The effect of peroxysome proliferator-activated receptor-γ ligands on in vitro and in vivo models of COPD. Eur. Respir. J. 2013, 43, 409–420. [Google Scholar]
- Prestera, T.; Talalay, P.; Alam, J.; Ahn, Y.I.; Lee, P.J.; Choi, A.M. Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: Regulation by upstream antioxidant-responsive elements (ARE). Mol. Med. 1995, 1, 827–837. [Google Scholar]
- Bonelli, M.; Savitskaya, A.; Steiner, C.W.; Rath, E.; Bilban, M.; Wagner, O.; Bach, F.H.; Smolen, J.S.; Scheinecker, C. Heme oxygenase-1 end-products carbon monoxide and biliverdin ameliorate murine collagen induced arthritis. Clin. Exp. Rheumatol. 2012, 30, 73–78. [Google Scholar]
- Patel, R.; Albadawi, H.; Steudel, W.; Hashmi, F.F.; Kang, J.; Yoo, H.J.; Watkins, M.T. Inhalation of carbon monoxide reduces skeletal muscle injury after hind limb ischemia-reperfusion injury in mice. Am. J. Surg. 2012, 203, 488–495. [Google Scholar] [PubMed] [Green Version]
- Liu, Y.; Zhu, B.; Wang, X.; Luo, L.; Li, P.; Paty, D.W.; Cynader, M.S. Bilirubin as a potent antioxidant suppresses experimental autoimmune encephalomyelitis: Implications for the role of oxidative stress in the development of multiple sclerosis. J. Neuroimmunol. 2003, 139, 27–35. [Google Scholar] [PubMed]
- Sawada, K.; Ohnishi, K.; Kosaka, T.; Chikano, S.; Egashira, A.; Okui, M.; Shintani, S.; Wada, M.; Nakasho, K.; Shimoyama, T. Exacerbated autoimmune hepatitis successfully treated with leukocytapheresis and bilirubin adsorption therapy. J. Gastroenterol. 1997, 32, 689–695. [Google Scholar]
- Kawamura, K.; Ishikawa, K.; Wada, Y.; Kimura, S.; Matsumoto, H.; Kohro, T.; Itabe, H.; Kodama, T.; Maruyama, Y. Bilirubin from heme oxygenase-1 attenuates vascular endothelial activation and dysfunction. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 155–160. [Google Scholar]
- Lanone, S.; Bloc, S.; Foresti, R.; Almolki, A.; Taille, C.; Callebert, J.; Conti, M.; Goven, D.; Aubier, M.; Dureuil, B.; et al. Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: Implications for protection against endotoxic shock in rats. FASEB J. 2005, 19, 1890–1892. [Google Scholar] [PubMed] [Green Version]
- Jun, Y.J.; Lee, M.; Shin, T.; Yoon, N.; Kim, J.H.; Kim, H.R. Eckol enhances heme oxygenase-1 expression through activation of Nrf2/JNK pathway in HepG2 cells. Molecules 2014, 19, 15638–15652. [Google Scholar]
- Lee, J.; Kim, S. Upregulation of heme oxygenase-1 expression by dehydrodiconiferyl alcohol (DHCA) through the AMPK-Nrf2 dependent pathway. Toxicol. Appl. Pharmacol. 2014, 281, 87–100. [Google Scholar]
- Choi, Y.H. The cytoprotective effect of isorhamnetin against oxidative stress is mediated by the upregulation of the Nrf2-dependent HO-1 expression in C2C12 myoblasts through scavenging reactive oxygen species and ERK inactivation. Gen. Physiol. Biophys. 2016, 35, 145–154. [Google Scholar]
- Zipper, L.M.; Mulcahy, R.T. ERK activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 Cells. Toxicol. Sci. 2003, 73, 124–134. [Google Scholar]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [PubMed]
- Sekine, H.; Okazaki, K.; Ota, N.; Shima, H.; Katoh, Y.; Suzuki, N.; Igarashi, K.; Ito, M.; Motohashi, H.; Yamamoto, M. The mediator subunit MED16 transduces NRF2-activating signals into antioxidant gene expression. Mol. Cell. Biol. 2015, 36, 407–420. [Google Scholar] [PubMed] [Green Version]
- Ziady, A.G.; Sokolow, A.; Shank, S.; Corey, D.; Myers, R.; Plafker, S.; Kelley, T.J. Interaction with CREB binding protein modulates the activities of Nrf2 and NF-κB in cystic fibrosis airway epithelial cells. Am. J. Phys. Lung Cell. Mol. Physiol. 2012, 302, L1221–L1231. [Google Scholar]
- Sharma, D.; Fondell, J.D. Ordered recruitment of histone acetyltransferases and the TRAP/Mediator complex to thyroid hormone-responsive promoters in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 7934–7939. [Google Scholar] [PubMed] [Green Version]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [PubMed] [Green Version]
- Okuda, Y.; Sakoda, S.; Bernard, C.C.; Fujimura, H.; Saeki, Y.; Kishimoto, T.; Yanagihara, T. IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int. Immunol. 1998, 10, 703–708. [Google Scholar]
- Sutton, C.; Brereton, C.; Keogh, B.; Mills, K.H.G.; Lavelle, E.C. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1685–1691. [Google Scholar]
- Johnson, D.A.; Amirahmadi, S.; Ward, C.; Fabry, Z.; Johnson, J.A. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol. Sci. 2010, 114, 237–246. [Google Scholar]
- Pareek, T.K.; Belkadi, A.; Kesavapany, S.; Zaremba, A.; Loh, S.L.; Bai, L.; Cohen, M.L.; Meyer, C.; Liby, K.T.; Miller, R.H.; et al. Triterpenoid modulation of IL-17 and Nrf-2 expression ameliorates neuroinflammation and promotes remyelination in autoimmune encephalomyelitis. Sci. Rep. 2011, 1, 201. [Google Scholar]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar]
- Pae, H.O.; Oh, G.S.; Lee, B.S.; Rim, J.S.; Kim, Y.M.; Chung, H.T. 3-Hydroxyanthranilic acid, one of L-tryptophan metabolies, inhibits monocyte chemoattractant protein-1 secretion and vascular cell adhesion molecule-1 expression via heme oxygenase-1 induction in human umbilical vein endothelial cells. Atherosclerosis 2006, 187, 274–284. [Google Scholar] [PubMed]
- Satoh, H.; Moriguchi, T.; Taguchi, K.; Takai, J.; Maher, J.M.; Suzuki, T.; Winnard, P.T., Jr.; Raman, V.; Ebina, M.; Nukiwa, T.; et al. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis 2010, 31, 1833–1843. [Google Scholar] [PubMed] [Green Version]
- Hiramoto, K.; Satoh, H.; Suzuki, T.; Moriguchi, T.; Pi, J.; Shimosegawa, T.; Yamamoto, M. Myeloid lineage-specific deletion of antioxidant system enhances tumor metastasis. Cancer Prev. Res. 2014, 7, 835–844. [Google Scholar]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [PubMed] [Green Version]
- Uruno, A.; Furusawa, Y.; Yagishita, Y.; Fukutomi, T.; Muramatsu, H.; Negishi, T.; Sugawara, A.; Kensler, T.W.; Yamamoto, M. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol. Cell. Biol. 2013, 33, 2996–3010. [Google Scholar] [PubMed] [Green Version]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Vølund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar]
- Shah, P.K. Inflammation, neointimal hyperplasia, and restenosis: As the leukocytes roll, the arteries thicken. Circulation 2003, 107, 2175–2177. [Google Scholar]
- Schiffrin, E.L. Beyond blood pressure: The endothelium and atherosclerosis progression. Am. J. Hypertens. 2002, 15, 115S–122S. [Google Scholar]
- Kim, J.H.; Choi, Y.K.; Lee, K.S.; Cho, D.H.; Baek, Y.Y.; Lee, D.K.; Ha, K.S.; Choe, J.; Wonc, M.H.; Jeoung, D.; et al. Functional dissection of Nrf2-dependent phase II genes in vascular inflammation and endotoxic injury using Keap1 siRNA. Free Radic. Biol. Med. 2012, 53, 629–640. [Google Scholar]
- Jin, W.; Wang, H.; Yan, W.; Xu, L.; Wang, X.; Zhao, X.; Yang, X.; Chen, G.; Ji, Y. Disruption of Nrf2 enhances upregulation of Nuclear Factor-κB activity, proinflammatory cytokines, and intercellular adhesion molecule-1 in the brain after traumatic brain injury. Med. Inflamm. 2008, 2008, 725174. [Google Scholar]
- Zakkar, M.; Heiden, K.V.; Luong, L.A.; Chaudhury, H.; Cuhlmann, S.; Hamdulay, S.S.; Krams, R.; Edirisinghe, I.; Rahman, I.; Carlsen, H.; et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1851–1857. [Google Scholar] [PubMed] [Green Version]
- Zakkar, M.; Chaudhury, H.; Sandvik, G.; Enesa, K.; Luong, L.A.; Cuhlmann, S.; Mason, J.C.; Krams, R.; Clark, A.R.; Haskard, D.O.; et al. Increased endothelial mitogen-activated protein kinase phosphatase-1 expression suppresses proinflammatory activation at sites that are resistant to atherosclerosis. Circ. Res. 2008, 103, 726–732. [Google Scholar] [PubMed] [Green Version]
- Yang, P.M.; Chen, H.Z.; Huang, Y.T.; Hsieh, C.W.; Wung, B.S. Lycopene inhibits NF-κB activation and adhesion molecule expression through Nrf2-mediated heme oxygenase-1 in endothelial cells. Int. J. Mol. Med. 2017, 39, 1533–1540. [Google Scholar]
- Koo, H.J.; Sohn, E.H.; Pyo, S.; Woo, H.G.; Park, D.W.; Ham, Y.M.; Jang, S.A.; Park, S.Y.; Kang, S.C. An ethanol root extract of Cynanchum wilfordii containing acetophenones suppresses the expression of VCAM-1 and ICAM-1 in TNF-α-stimulated human aortic smooth muscle cells through the NF-κB pathway. Int. J. Mol. Med. 2015, 35, 915–924. [Google Scholar] [PubMed] [Green Version]
- Sarah, A.; Struck, B.; Genrich, G.; Helm, O.; Sipos, B.; Sebens, S.; Schäfer, H. The crosstalk between Nrf2 and TGF-β1 in the epithelial-mesenchymal transition of pancreatic duct epithelial cells. PLoS ONE 2015, 10, e0132978. [Google Scholar]
- Coppini, R.; Santini, L.; Palandri, C.; Sartiani, L.; Cerbai, E.; Raimondi, L. Pharmacological inhibition of serine proteases to reduce cardiac inflammation and fibrosis in atrial fibrillation. Front. Pharmacol. 2019, 10, 1420. [Google Scholar] [PubMed]
- Ishii, Y.; Itoh, K.; Morishima, Y.; Kimura, T.; Kiwamoto, T.; Izuka, T.I.; Hegab, A.E.; Hosoya, T.; Nomura, A.; Sakamoto, T.; et al. Transcription factor Nrf2 plays a pivotal role in protection against Elastase-induced pulmonary inflammation and emphysema. J. Immunol. 2005, 175, 6968–6975. [Google Scholar]
- Jin, F.Y.; Nathan, C.; Radzioch, D.; Ding, A. Secretory leukocyte protease inhibitor a macrophage product induced by and antagonistic to bacterial lipopolysaccharide. Cell 1997, 88, 417–426. [Google Scholar]
- Sallenave, J.M.; Shulmann, J.; Crossley, J.; Jordana, M.; Gauldie, J. Regulation of secretory leukocyte proteinase inhibitor (SLPI) and elastase-specific inhibitor (ESI/elafin) in human airway epithelial cells by cytokines and neutrophilic enzymes. Am. J. Respir. Cell Mol. Biol. 1994, 11, 733–741. [Google Scholar]
- Meyer, M.; Kesic, M.J.; Clarke, J.; Ho, E.; Simmen, R.C.; Diaz-Sanchez, D.; Noah, T.L.; Jaspers, I. Sulforaphane induces SLPI secretion in the nasal mucosa. Respir. Med. 2012, 107, 472–475. [Google Scholar]
- Meyer, M.; Jaspers, I. Respiratory protease/antiprotease balance determines susceptibility to viral infection and can be modified by nutritional antioxidants. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L1189–L1201. [Google Scholar] [PubMed] [Green Version]
- Saw, C.L.L.; Yang, A.Y.; Huang, M.T.; Liu, Y.; Lee, J.H.; Khor, T.O.; Su, Z.Y.; Shu, L.; Lu, Y.; Conney, A.H.; et al. Nrf2 null enhances UVB-induced skin inflammation and extracellular matrix damages. Cell Biosci. 2014, 4, 39. [Google Scholar] [PubMed] [Green Version]
- Chaiprasongsuk, A.; Lohakul, J.; Soontrapa, K.; Sampattavanich, S.; Akarasereenont, P.; Panich, U. Activation of Nrf2 Reduces UVA-Mediated MMP-1 upregulation via MAPK/AP-1 signaling cascades: The photoprotective effects of Sulforaphane and Hispidulins. J. Pharmacol. Exp. Ther. 2017, 360, 388–398. [Google Scholar] [PubMed] [Green Version]
- Hyeon, S.; Lee, H.; Yang, Y.; Jeong, W. Nrf2 deficiency induces oxidative stress and promotes RANKL-induced osteoclast differentiation. Free Radic. Biol. Med. 2013, 65, 789–799. [Google Scholar] [PubMed]
- Kim, B.C.; Jeon, W.K.; Hong, H.Y.; Jeon, K.B.; Hahn, J.H.; Kim, Y.M.; Numazawa, S.; Yosida, T.; Park, E.H.; Lim, C.J. The anti-inflammatory activity of Phellinus linteus (Berk. & M.A. Curt.) is mediated through the PKCdelta/Nrf2/ARE signaling to upregulation of heme oxygenase-1. J. Ethnopharmacol. 2007, 113, 240–247. [Google Scholar] [PubMed]
- Lee, S.H.; Sohn, D.H.; Jin, X.Y.; Kim, S.W.; Choi, S.C.; Seo, G.S. 2’,4’,6’-tris(methoxymethoxy) chalcone protects against trinitrobenzene sulfonic acid-induced colitis and blocks tumor necrosis factor-alpha-induced intestinal epithelial inflammation via heme oxygenase 1-dependent and independent pathways. Biochem. Pharmacol. 2007, 74, 870–880. [Google Scholar]
- Mao, L.; Wang, H.; Qiao, L.; Wang, X. Disruption of Nrf2 enhances the upregulation of Nuclear Factor-kappaB activity, Tumor Necrosis Factor-α, and Matrix Metalloproteinase-9 after spinal cord injury in mice. Med. Inflamm. 2010, 2010, 238321. [Google Scholar]
- Cai, D.; Yin, S.; Yang, J.; Jiang, Q.; Cao, W. Histone deacetylase inhibition activates Nrf2 and protects against osteoarthritis. Art. Res. Ther. 2015, 17, 269. [Google Scholar]
- Long, M.; Vega, M.R.; Wen, Q.; Bharara, M.; Jiang, T.; Zhang, R.; Zhou, S.; Wong, P.K.; Wondrak, G.T.; Zheng, H.; et al. An essential role of NRF2 in diabetic wound healing. Diabetes 2016, 65, 780–793. [Google Scholar]
- Nicholson, A.C.; Febbraio, M.; Han, J.; Silverstein, R.L.; Hajjar, D.P. CD36 in atherosclerosis: The role of a class B macrophage scavenger receptor. Ann. N. Y. Acad. Sci. 2000, 902, 128–131. [Google Scholar]
- Yoshida, H.; Quehenberger, O.; Kondratenko, N.; Green, S.; Steinberg, D. Minimally oxidized low-density lipoprotein increases expression of scavenger receptor A, CD36, and macrosialin in resident mouse peritoneal macrophages. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 794–802. [Google Scholar] [PubMed] [Green Version]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages. Circul. Res. 2004, 94, 609–616. [Google Scholar]
- Takabe, W.; Warabi, E.; Noguchi, N. Anti-atherogenic effect of laminar shear stress via Nrf2 activation. Antioxid. Redox Signal. 2011, 15, 1415–1426. [Google Scholar]
- Barajas, B.; Che, N.; Yin, F.; Rowshanrad, A.; Orozco, L.D.; Gong, K.W.; Wang, X.; Castellani, L.W.; Reue, K.; Lusis, A.J.; et al. NF-E2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 58–66. [Google Scholar] [PubMed] [Green Version]
- Sussan, T.E.; Jun, J.; Thimmulappa, R.; Bedja, D.; Antero, M.; Gabrielson, K.L.; Polotsky, V.Y.; Biswal, S. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS ONE 2008, 3, e3791. [Google Scholar]
- Zhao, X.; Sun, G.; Ting, S.M.; Song, S.; Zhang, J.; Edwards, N.J.; Aronowski, J. Cleaning up after ICH: The role of Nrf2 in modulating microglia function and hematoma clearance. J. Neurochem. 2015, 133, 144–152. [Google Scholar]
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852. [Google Scholar]
- Seibert, K.; Zhang, Y.; Leahy, K.; Hauser, S.; Masferrer, J.; Perkins, W.; Lee, L.; Isakson, P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc. Natl. Acad. Sci. USA 1994, 91, 12013–12017. [Google Scholar]
- Kundu, J.K.; Surh, Y.J. Nrf2-Keap1 signaling as a potential target for chemoprevention of inflammation-associated carcinogenesis. Pharm. Res. 2010, 27, 999–1013. [Google Scholar]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar]
- Itoh, K.; Mochizuki, M.; Ishii, Y.; Ishii, T.; Shibata, T.; Kawamoto, Y.; Kelly, V.; Sekizawa, K.; Uchida, K.; Yamamoto, M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Δ12,14-prostaglandin J2. Mol. Cell. Biol. 2004, 24, 36–45. [Google Scholar] [PubMed] [Green Version]
- Mochizuki, M.; Ishii, Y.; Itoh, K.; Iizuka, T.; Morishima, Y.; Kimura, T.; Kiwamoto, T.; Matsuno, Y.; Hegab, A.E.; Nomura, A.; et al. Role of 15-deoxy-Δ12,14-prostaglandin J2 and Nrf2 pathways in protection against acute lung injury. Am. J. Respir. Crit. Care Med. 2005, 171, 1260–1266. [Google Scholar] [PubMed]
- Gong, P.; Stewart, D.; Hu, B.; Li, N.; Cook, J.; Nel, A.; Alam, J. Activation of the mouse heme oxygenase-1 gene by 15-deoxy-Δ12,14-prostaglandin J2 is mediated by the stress response elements and transcription factor Nrf2. Antioxid. Redox Signal. 2002, 4, 249–257. [Google Scholar]
- Levonen, A.L.; Landar, A.; Ramachandran, A.; Ceaser, E.K.; Dickinson, D.A.; Zanoni, G.; Morrow, J.D.; Darley-Usmar, V.M. Cellular mechanisms of redox cell signaling: Role of cysteine modification in controlling antioxidant defenses in response to electrophilic lipid oxidation products. Biochem. J. 2004, 378, 373–382. [Google Scholar]
- Hsieh, C.Y.; Hsiao, H.Y.; Wu, W.Y.; Liu, C.A.; Tsai, Y.C.; Chao, Y.J.; Wang, D.L.; Hsieh, H.-J. Regulation of shear-induced nuclear translocation of the Nrf2 transcription factor in endothelial cells. J. Biomed. Sci. 2009, 16, 12. [Google Scholar]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar]
- Lin, W.; Wu, R.T.; Wu, T.; Khor, T.O.; Wang, H.; Kong, A.N. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochem. Pharmacol. 2008, 76, 967–973. [Google Scholar]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome- activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar]
- Liu, X.; Zhang, X.; Ding, Y.; Zhou, W.; Tao, L.; Lu, P.; Wang, Y.; Hu, R. Nuclear factor E2-related factor-2 negatively regulates NLRP3 inflammasome activity by inhibiting reactive oxygen species- induced NLRP3 priming. Antioxid. Redox Signal. 2017, 26, 28–43. [Google Scholar] [PubMed] [Green Version]
- Li, H.Y.; Zhong, Y.F.; Wu, S.Y.; Shi, N. NF-E2 related factor 2 activation and heme oxygenase-1 induction by tert-butylhydroquinone protect against deltamethrin-mediated oxidative stress in PC12 cells. Chem. Res. Toxicol. 2007, 20, 1242–1251. [Google Scholar] [PubMed]
- Tsai, P.Y.; Ka, S.M.; Chang, J.M.; Chen, H.C.; Shui, H.A.; Li, C.Y.; Hua, K.F.; Chang, W.L.; Huang, J.J.; Yang, S.S.; et al. Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radic. Biol. Med. 2011, 51, 744–754. [Google Scholar] [PubMed]
- Ka, S.M.; Lin, J.C.; Lin, T.J.; Liu, F.C.; Chao, L.K.; Ho, C.L.; Yeh, L.T.; Sytwu, H.K.; Hua, K.F.; Chen, A. Citral alleviates an accelerated and severe lupus nephritis model by inhibiting the activation signal of NLRP3 inflammasome and enhancing Nrf2 activation. Arth. Res. Ther. 2015, 17, 331. [Google Scholar]
- Liu, X.; Wang, T.; Cai, L.; Qi, J.; Zhang, P.; Li, Y. Biochanin A protects lipopolysaccharide/ D-galactosamine-induced acute liver injury inmice by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation. Int. Immunopharmacol. 2016, 38, 324–331. [Google Scholar] [PubMed]
- Liu, Q.; Lv, H.; Wen, Z.; Ci, X.; Peng, L. Isoliquiritigenin activates Nuclear Factor Erythroid-2 Related Factor 2 to suppress the NOD-Like Receptor Protein 3 inflammasome and inhibits the NF-κB pathway in macrophages and in acute lung injury. Front. Immunol. 2017, 8, 1518. [Google Scholar] [PubMed]
- He, L.; Peng, X.; Zhu, J.; Chen, X.; Liu, H.; Tang, C.; Dong, Z.; Liu, F.; Peng, Y. Mangiferin attenuate sepsis-induced acute kidney injury via antioxidant and anti-inflammatory effects. Am. J. Nephrol. 2014, 40, 441–450. [Google Scholar]
- Arioz, B.I.; Tastan, B.; Tarakcioglu, E.; Tufekci, K.U.; Olcum, M.; Ersoy, N.; Bagriyanik, A.; Genc, K.; Genc, S. Melatonin attenuates LPS-induced acute depressive-like behaviors and microglial NLRP3 inflammasome activation through the SIRT1/Nrf2 pathway. Front. Immunol. 2019, 10, 1511. [Google Scholar]
- Greaney, A.J.; Maier, N.K.; Leppla, S.H.; Moayeri, M. Sulforaphane inhibits multiple inflammasomes through an Nrf2-independent mechanism. J. Leukoc. Biol. 2016, 99, 189–199. [Google Scholar]
- An, Y.W.; Jhang, K.A.; Woo, S.Y.; Kang, J.L.; Chong, Y.H. Sulforaphane exerts its anti-inflammatory effect against amyloid-β peptide via STAT-1 dephosphorylation and activation of Nrf2/HO-1 cascade in human THP-1 macrophages. Neurobiol. Aging 2016, 38, 1–10. [Google Scholar]
- Lv, H.; Liu, Q.; Wen, Z.; Feng, H.; Deng, X.; Ci, X. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3β-Nrf2 signal axis. Redox Biol. 2017, 12, 311–324. [Google Scholar]
- Dong, Z.; Shang, H.; Chen, Y.Q.; Pan, L.L.; Bhatia, M.; Sun, J. Sulforaphane protects pancreatic acinar cell injury by modulating Nrf2- mediated oxidative stress and NLRP3 inflammatory pathway. Oxid. Med. Cell Longev. 2016, 2016, 7864150. [Google Scholar]
- Zhao, C.; Gillette, D.D.; Li, X.; Zhang, Z.; Wen, H. Nuclear factor E2-related factor-2 (Nrf2) is required for NLRP3 and AIM2 inflammasome activation. J. Biol. Chem. 2014, 289, 17020–17029. [Google Scholar] [PubMed] [Green Version]
- Freigang, S.; Ampenberger, F.; Spohn, G.; Heer, S.; Shamshiev, A.T.; Kisielow, J.; Hersberger, M.; Yamamoto, M.; Bachmann, M.F.; Kopf, M. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 2011, 41, 2040–2051. [Google Scholar] [PubMed]
- Jhang, J.J.; Cheng, Y.T.; Ho, C.Y.; Yen, G.C. Monosodium urate crystals trigger Nrf2- and heme oxygenase-1-dependent inflammation in THP-1 cells. Cell Mol. Immunol. 2015, 12, 424–434. [Google Scholar] [PubMed] [Green Version]
- Reddy, N.M.; Suryanarayana, V.; Kalvakolanu, D.V.; Yamamoto, M.; Kensler, T.W.; Hassoun, P.M.; Kleeberger, S.R.; Reddy, S.P. Innate immunity against bacterial infection following hyperoxia exposure is impaired in NRF2-deficient mice. J. Immunol. 2009, 183, 4601–4608. [Google Scholar]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar]
- Schjerning, A.; McGettigan, P.; Gislason, G. Cardiovascular effects and safety of (non-aspirin) NSAIDs. Nat. Rev. Cardiol. 2020, 17, 574–584. [Google Scholar]
- Jian, Z.; Tang, L.; Yi, X.; Liu, B.; Zhang, Q.; Zhu, G.; Wang, G.; Gao, T.; Li, C. Aspirin induces Nrf2-mediated transcriptional activation of haem oxygenase-1 in protection of human melanocytes from H2O2-induced oxidative stress. J. Cell Mol. Med. 2016, 20, 1307–1318. [Google Scholar]
- Wang, W.; Chen, S.; Zhou, Z.; Dai, H.; Wang, H.; Li, Y.; Zhou, K.; Shen, Z.; Guo, Y.; Liu, C.; et al. Aspirin suppresses neuronal apoptosis, reduces tissue inflammation, and restrains astrocyte activation by activating the Nrf2/HO-1 signaling pathway. Neuroreport 2018, 29, 524–531. [Google Scholar]
- Wang, J.S.; Ho, F.M.; Kang, H.C.; Lin, W.W.; Huang, K.C. Celecoxib induces heme oxygenase-1 expression in macrophages and vascular smooth muscle cells via ROS-dependent signaling pathway. Naunyn. Schmiedebergs Arch. Pharmacol. 2011, 383, 159–168. [Google Scholar] [PubMed]
- Al-Rashed, F.; Calay, D.; Lang, M.; Thornton, C.C.; Bauer, A.; Kiprianos, A.; Haskard, D.O.; Seneviratne, A.; Boyle, J.J.; Schönthal, A.H.; et al. Celecoxib exerts protective effects in the vascular endothelium via COX-2-independent activation of AMPK-CREB-Nrf2 signalling. Sci. Rep. 2018, 8, 6271. [Google Scholar] [PubMed] [Green Version]
- Bao, S.; Nie, X.; Ou, R.; Wang, C.; Ku, P.; Li, K. Effects of diclofenac on the expression of Nrf2 and its downstream target genes in mosquito fish (Gambusia affinis). Aquat. Toxicol. 2017, 188, 43–53. [Google Scholar] [PubMed] [Green Version]
- Yoshinaga, N.; Arimura, N.; Otsuka, H.; Kawahara, K.; Hashiguchi, T.; Maruyama, I.; Sakamoto, T. NSAIDs inhibit neovascularization of choroid through HO-1-dependent pathway. Lab. Investig. 2011, 91, 1277–1290. [Google Scholar] [PubMed]
- Lee, H.J.; Han, Y.M.; Kim, E.H.; Kim, Y.J.; Hahm, K.B. A possible involvement of Nrf2-mediated heme oxygenase-1 up-regulation in protective effect of the proton pump inhibitor pantoprazole against indomethacin-induced gastric damage in rats. BMC Gastroenterol. 2012, 12, 143. [Google Scholar]
- Dunlap, T.; Chandrasena, R.E.; Wang, Z.; Sinha, V.; Thatcher, G.R.J. Quinone formation as a chemoprevention strategy for hybrid drugs: Balancing cytotoxicity and cytoprotection. Chem. Res. Toxicol. 2007, 20, 1903–1912. [Google Scholar] [PubMed]
- Gao, J.; Kashfi, K.; Liu, X.; Rigas, B. NO-donating aspirin induces phase II enzymes in vitro and in vivo. Carcinogenesis 2006, 27, 803–810. [Google Scholar]
- Hulsman, N.; Medema, J.P.; Bos, C.; Jongejan, A.; Leurs, R.; Smit, M.J.; de Esch, I.J.; Richel, D.; Wijtmans, M. Chemical insights in the concept of hybrid drugs: The antitumor effect of nitric oxide-donating aspirin involves a quinone methide but not nitric oxide nor aspirin. J. Med. Chem. 2007, 50, 2424–2431. [Google Scholar]
- Dunlap, T.; Piyankarage, S.C.; Wijewickrama, G.T.; Abdul-Hay, S.; Vanni, M.; Litosh, V.; Luo, J.; Thatcher, G.R.J. Quinone-induced activation of Keap1/Nrf2 signaling by aspirin prodrugs masquerading as nitric oxide. Chem. Res. Toxicol. 2012, 25, 2725–2736. [Google Scholar]
- Zhu, H.; Jia, Z.; Li, Y.R. Nrf2 signaling in macrophages. React. Oxy. Sp. (Apex) 2016, 2, 417–420. [Google Scholar]
- Arellano-Buendía, A.S.; Tostado-González, M.; García-Arroyo, F.E.; Cristóbal-García, M.; Loredo-Mendoza, M.L.; Tapia, E.; Sánchez-Lozada, L.G.; Osorio-Alonso, H. Anti-Inflammatory therapy modulates Nrf2-Keap1 in kidney from rats with diabetes. Oxid. Med. Cell. Longev. 2016, 2016, 4693801. [Google Scholar] [PubMed] [Green Version]
- Cheng, X.; Gao, D.X.; Song, J.J.; Ren, F.Z.; Mao, X.Y. Casein glycomacropeptide hydrolysate exerts cytoprotection against H2O2-induced oxidative stress in RAW 264.7 macrophages via ROS-dependent heme oxygenase-1 expression. RSC Adv. 2015, 5, 4511–4523. [Google Scholar]
- Li, T.; Chen, B.; Du, M.; Song, J.; Cheng, X.; Wang, X.; Mao, X. Casein Glycomacropeptide hydrolysates exert cytoprotective effect against cellular oxidative stress by up-regulating HO-1 expression in HepG2 cells. Nutrients 2017, 9, 31. [Google Scholar]
- Quinti, L.; Naidu, S.D.; Träger, U.; Chen, X.; Kegel-Gleason, K.; Llères, D.; Connolly, C.; Chopra, V.; Low, C.; Moniot, S.; et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc. Natl Acad. Sci. USA 2017, 114, E4676–E4685. [Google Scholar] [PubMed] [Green Version]
- Quinti, L.; Casale, M.; Moniot, S.; Pais, T.F.; Van Kanegan, M.J.; Kaltenbach, L.S.; Pallos, J.; Lim, R.G.; Naidu, S.D.; Runne, H.; et al. SIRT2- and NRF2-targeting thiazole- containing compound with therapeutic activity in Huntington’s disease models. Cell Chem. Biol. 2016, 23, 849–861. [Google Scholar]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar]
- Gold, R.; Linker, R.A.; Stangel, M. Fumaric acid and its esters: An emerging treatment for multiple sclerosis with antioxidative mechanism of action. Clin. Immunol. 2012, 142, 44–48. [Google Scholar]
- Mrowietz, U.; Christophers, E.; Altmeyer, P. Treatment of psoriasis with fumaric acid esters: Results of a prospective multicentre study. German multicentre study. Br. J. Dermatol. 1998, 138, 456–460. [Google Scholar]
- Ma, Y.; Kosinska-Cagnazzo, A.; Kerr, W.L.; Amarowicz, R.; Swanson, R.B.; Pegg, R.B. Separation and characterization of soluble esterified and glycoside-bound phenolic compounds in dry-blanched peanut skins by liquid chromatography-electrospray ionization mass spectrometry. J. Agric. Food Chem. 2014, 62, 11488–11504. [Google Scholar]
- Rose, P.; Won, Y.K.; Ong, C.N.; Whiteman, M. Beta-phenylethyl and 8-methylsulphinyloctyl isothiocyanates, constituents of watercress, suppress LPS induced production of nitric oxide and prostaglandin E2 in RAW 264.7 macrophages. Nitric Oxide 2005, 12, 237–243. [Google Scholar] [PubMed]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [PubMed] [Green Version]
- Kim, J.S.; Oh, J.M.; Choi, H.; Kim, S.W.; Kim, S.W.; Kim, B.G.; Cho, J.H.; Lee, J.; Lee, D.C. Activation of the Nrf2/HO-1 pathway by curcumin inhibits oxidative stress in human nasal fibroblasts exposed to urban particulate matter. BMC Comp. Med. Ther. 2020, 20, 101. [Google Scholar]
- Jiang, C.; Luo, P.; Li, X.; Liu, P.; Li, Y.; Xu, J. Nrf2/ARE is a key pathway for curcumin-mediated protection of TMJ chondrocytes from oxidative stress and inflammation. Cell Stress Chaperones 2020, 25, 395–406. [Google Scholar]
- Kim, A.N.; Jeon, W.K.; Lee, J.J.; Kim, B.C. Up-regulation of heme oxygenase-1 expression through CaMKII-ERK1/2-Nrf2 signaling mediates the anti-inflammatory effect of bisdemethoxycurcumin in LPS-stimulated macrophages. Free Radic. Biol. Med. 2010, 49, 323–331. [Google Scholar]
- Pugazhenthi, S.; Akhov, L.; Selvaraj, G.; Wang, M.; Alam, J. Regulation of heme oxygenase-1 expression by demethoxy curcuminoids through Nrf2 by a PI3-kinase/Akt-mediated pathway in mouse beta-cells. Am. J. Phys. Endocrinol. Metabol. 2007, 293, E645–E655. [Google Scholar]
- Li, J.; Sapper, T.N.; Mah, E.; Rudraiah, S.; Schill, K.E.; Chitchumroonchokchai, C.; Moller, M.V.; McDonald, J.D.; Rohrer, P.R.; Manautou, J.E.; et al. Green tea extract provides extensive Nrf2-independent protection against lipid accumulation and NFkappaB pro-inflammatory responses during nonalcoholic steatohepatitis in mice fed a high-fat diet. Mol. Nutr. Food Res. 2016, 60, 858–870. [Google Scholar]
- Zhu, G.F.; Guo, H.J.; Huang, Y.; Wu, C.T.; Zhang, X.F. Eriodictyol, a plant flavonoid, attenuates LPS-induced acute lung injury through its antioxidative and anti-inflammatory activity. Exp. Ther. Med. 2016, 10, 2259–2266. [Google Scholar]
- Li, C.Z.; Jin, H.H.; Sun, H.X.; Zhang, Z.Z.; Zheng, J.X.; Li, S.H.; Han, S.H. Eriodictyol attenuates cisplatin-induced kidney injury by inhibiting oxidative stress and inflammation. Eur. J. Pharmacol. 2016, 772, 124–130. [Google Scholar]
- Boyanapalli, S.S.; Paredes-Gonzalez, X.; Fuentes, F.; Zhang, C.; Guo, Y.; Pung, D.; Saw, C.L.; Kong, A.N. Nrf2 knockout attenuates the anti-inflammatory effects of phenethyl isothiocyanate and curcumin. Chem. Res. Toxicol. 2016, 27, 2036–2043. [Google Scholar]
- Leonardo, C.C.; Dore, S. Dietary flavonoids are neuroprotective through Nrf2-coordinated induction of endogenous cytoprotective proteins. Nutr. Neurosci. 2016, 14, 226–236. [Google Scholar]
- Wang, Y.; Wang, B.; Du, F.; Su, X.; Sun, G.; Zhou, G.; Bian, X.; Liu, N. Epigallocatechin-3-Gallate attenuates oxidative stress and inflammation in obstructive nephropathy via NF-κB and Nrf2/HO-1 signalling pathway regulation. Basic Clin. Pharmacol. Tocicol. 2015, 117, 164–172. [Google Scholar]
- Kanlaya, R.; Khamchun, S.; Kapincharanon, C.; Thongboonkerd, V. Protective effect of epigallocatechin-3-gallate (EGCG) via Nrf2 pathway against oxalate-induced epithelial mesenchymal transition (EMT) of renal tubular cells. Sci. Rep. 2016, 6, 30233. [Google Scholar] [PubMed]
- Shanmugam, T.; Selvaraj, M.; Poomalai, S. Epigallocatechin gallate potentially abrogates fluoride induced lung oxidative stress, inflammation via Nrf2/Keap1 signaling pathway in rats: An in-vivo and in-silico study. Int. Immunopharmacol. 2016, 39, 128–139. [Google Scholar]
- Sun, W.; Liu, X.; Zhang, H.; Song, Y.; Li, T.; Liu, X.; Liu, Y.; Guo, L.; Wang, F.; Yang, T.; et al. Epigallocatechin gallate upregulates NRF2 to prevent diabetic nephropathy via disabling KEAP1. Free Radic. Biol. Med. 2017, 108, 840–857. [Google Scholar]
- Ruiz-Miyazawa, K.W.; Pinho-Ribeiro, F.A.; Borghi, S.M.; Staurengo-Ferrari, L.; Fattori, V.; Amaral, F.A.; Teixeira, M.M.; Alves-Filho, J.C.; Cunha, T.M.; Cunha, F.Q.; et al. Hesperidin methylchalcone suppresses experimental gout arthritis in mice by inhibiting NF-kappaB activation. J. Agric. Food Chem. 2018, 66, 6269–6280. [Google Scholar]
- Zhang, Y.; Liu, B.; Chen, X.; Zhang, N.; Li, G.; Zhang, L.H.; Tan, L.Y. Naringenin ameliorates behavioral dysfunction and neurological deficits in a d-galactose-induced aging mouse model through activation of PI3K/Akt/Nrf2 pathway. Rejuvenation Res. 2017, 20, 462–472. [Google Scholar]
- Calixto-Campos, C.; Corrêa, M.P.; Carvalho, T.T.; Zarpelon, A.C.; Hohmann, M.S.N.; Rossaneis, A.C.; Coelho-Silva, L.; Pavanelli, W.R.; Pinge-Filho, P.; Crespigio, J.; et al. Quercetin reduces ehrlich tumor-induced cancer pain in mice. Anal. Cell. Pathol. (Amst.) 2015, 2015, 285708. [Google Scholar]
- Borghi, S.M.; Mizokami, S.S.; Pinho-Ribeiro, F.A.; Fattori, V.; Crespigio, J.; Clemente-Napimoga, J.T.; Napimoga, M.H.; Pitol, D.L.; Issa, J.P.M.; Fukada, S.Y.; et al. The flavonoid quercetin inhibits titanium dioxide (TiO2)-induced chronic arthritis in mice. J. Nutr. Biochem. 2017, 53, 81–95. [Google Scholar]
- Anantharaju, P.G.; Gowda, P.C.; Vimalambike, M.G.; Madhunapantula, S.V. An overview on the role of dietary phenolics for the treatment of cancers. Nutr. J. 2016, 15, 99. [Google Scholar]
- Rong, H.; Liang, Y.; Niu, Y. Rosmarinic acid attenuates beta-amyloid-induced oxidative stress via Akt/GSK-3beta/Fyn-mediated Nrf2 activation in PC12 cells. Free Radic. Biol. Med. 2018, 120, 114–123. [Google Scholar] [PubMed]
- Kim, K.H.; Sadikot, R.T.; Joo, M. Therapeutic effect of ent-kaur-16-en-19-oic acid on neutrophilic lung inflammation and sepsis is mediated by Nrf2. Biochem. Biophys. Res. Commun. 2016, 474, 534–540. [Google Scholar] [PubMed]
- Kim, Y.M.; Kim, H.J.; Chang, K.C. Glycyrrhizin reduces HMGB1 secretion in lipopolysaccharide-activated RAW 264.7 cells and endotoxemic mice by p38/Nrf2-dependent induction of HO-1. Int. Immunopharmacol. 2015, 26, 112–118. [Google Scholar] [PubMed]
Figure 1.
A surface presentation of the Kelch domain (carton) with peptide from Neh2 domain of Nrf2 (sticks in mesh) from crystal structures: 2FLU.
Figure 1.
A surface presentation of the Kelch domain (carton) with peptide from Neh2 domain of Nrf2 (sticks in mesh) from crystal structures: 2FLU.

Figure 2.
Under normal homeostatic conditions, Keap1 homodimerizes through the N-terminal BTB domain and binds to the cullin-based (Cul3) E3 ligase, forming Keap1-Cul3-RBX1 (Ring box protein-1) E3 ligase complex, leading to Nrf2 ubiquitination and degradation. Under stress (electrophiles or ROS or endoplasmic reticulum (ER) stress) conditions, Nrf2 is released from Keap1-Cul3-RBX1 complex and translocates into the nucleus wherein it heterodimerizes with small Maf proteins (sMaf) and binds to the antioxidant response elements (AREs), leading to the transcription of ARE-driven genes.
Figure 2.
Under normal homeostatic conditions, Keap1 homodimerizes through the N-terminal BTB domain and binds to the cullin-based (Cul3) E3 ligase, forming Keap1-Cul3-RBX1 (Ring box protein-1) E3 ligase complex, leading to Nrf2 ubiquitination and degradation. Under stress (electrophiles or ROS or endoplasmic reticulum (ER) stress) conditions, Nrf2 is released from Keap1-Cul3-RBX1 complex and translocates into the nucleus wherein it heterodimerizes with small Maf proteins (sMaf) and binds to the antioxidant response elements (AREs), leading to the transcription of ARE-driven genes.

Figure 3.
Interplay of Nrf2/HO-1 axis and NF-κB in inflammation. Upon TLR signaling, NF-κB is liberated from IκB, by phosphorylation by the IKK complex. NF-κB then translocates into the nucleus and induces the expression of proinflammatory cytokines, CAMs and other molecules. Nrf2 induces increase in the cellular HO-1 expression and inhibits oxidative stress-mediated NF-κB activation and blocks the degradation of IκB-α. In addition, Nrf2 negatively regulates the IκB-α proteasomal degradation and nuclear translocation of NF-κB.
Figure 3.
Interplay of Nrf2/HO-1 axis and NF-κB in inflammation. Upon TLR signaling, NF-κB is liberated from IκB, by phosphorylation by the IKK complex. NF-κB then translocates into the nucleus and induces the expression of proinflammatory cytokines, CAMs and other molecules. Nrf2 induces increase in the cellular HO-1 expression and inhibits oxidative stress-mediated NF-κB activation and blocks the degradation of IκB-α. In addition, Nrf2 negatively regulates the IκB-α proteasomal degradation and nuclear translocation of NF-κB.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. https://doi.org/10.3390/molecules25225474
AMA Style
Saha S, Buttari B, Panieri E, Profumo E, Saso L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules. 2020; 25(22):5474. https://doi.org/10.3390/molecules25225474
Chicago/Turabian StyleSaha, Sarmistha, Brigitta Buttari, Emiliano Panieri, Elisabetta Profumo, and Luciano Saso. 2020. "An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation" Molecules 25, no. 22: 5474. https://doi.org/10.3390/molecules25225474