Casticin Induces DNA Damage and Affects DNA Repair Associated Protein Expression in Human Lung Cancer A549 Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
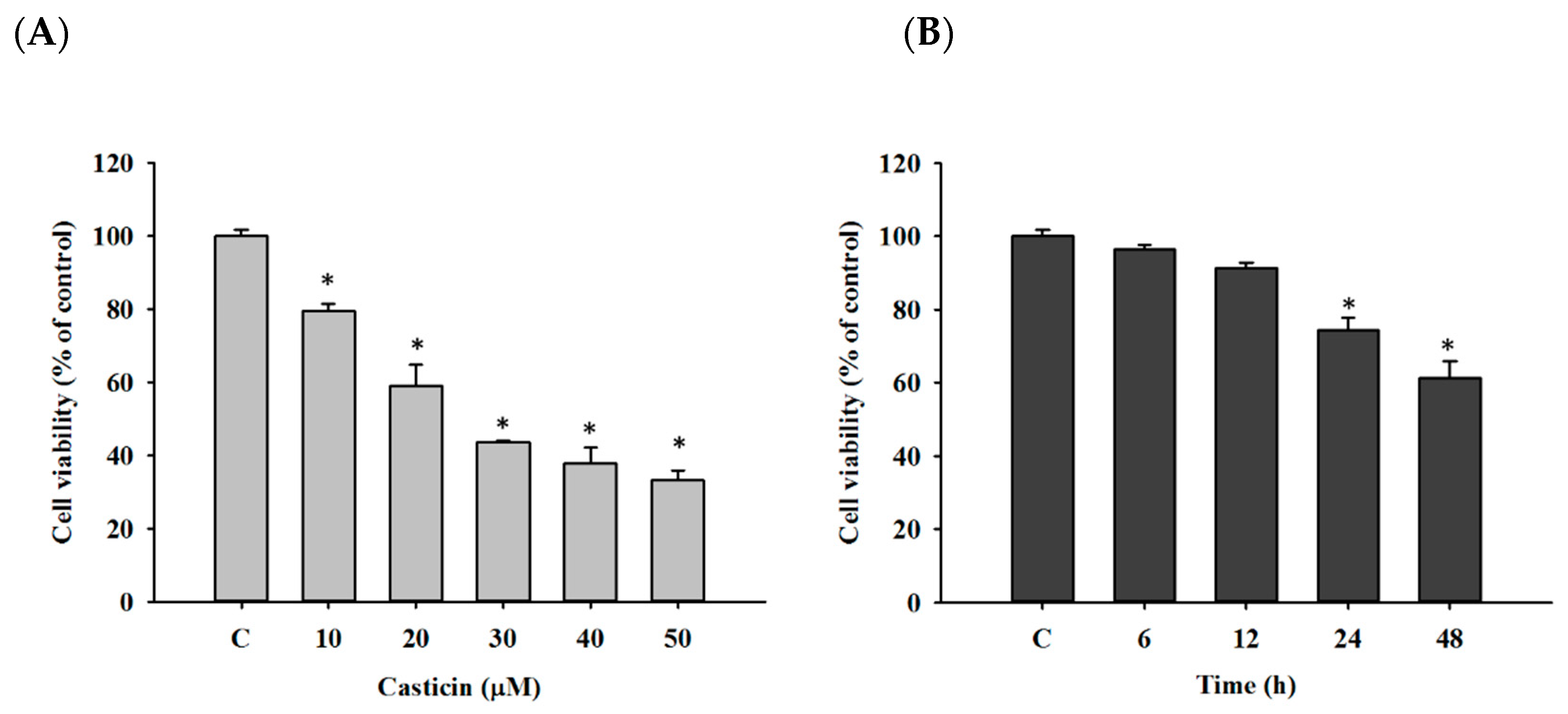
2.1. Casticin Decreased Viable Cell Number of A549 Cells
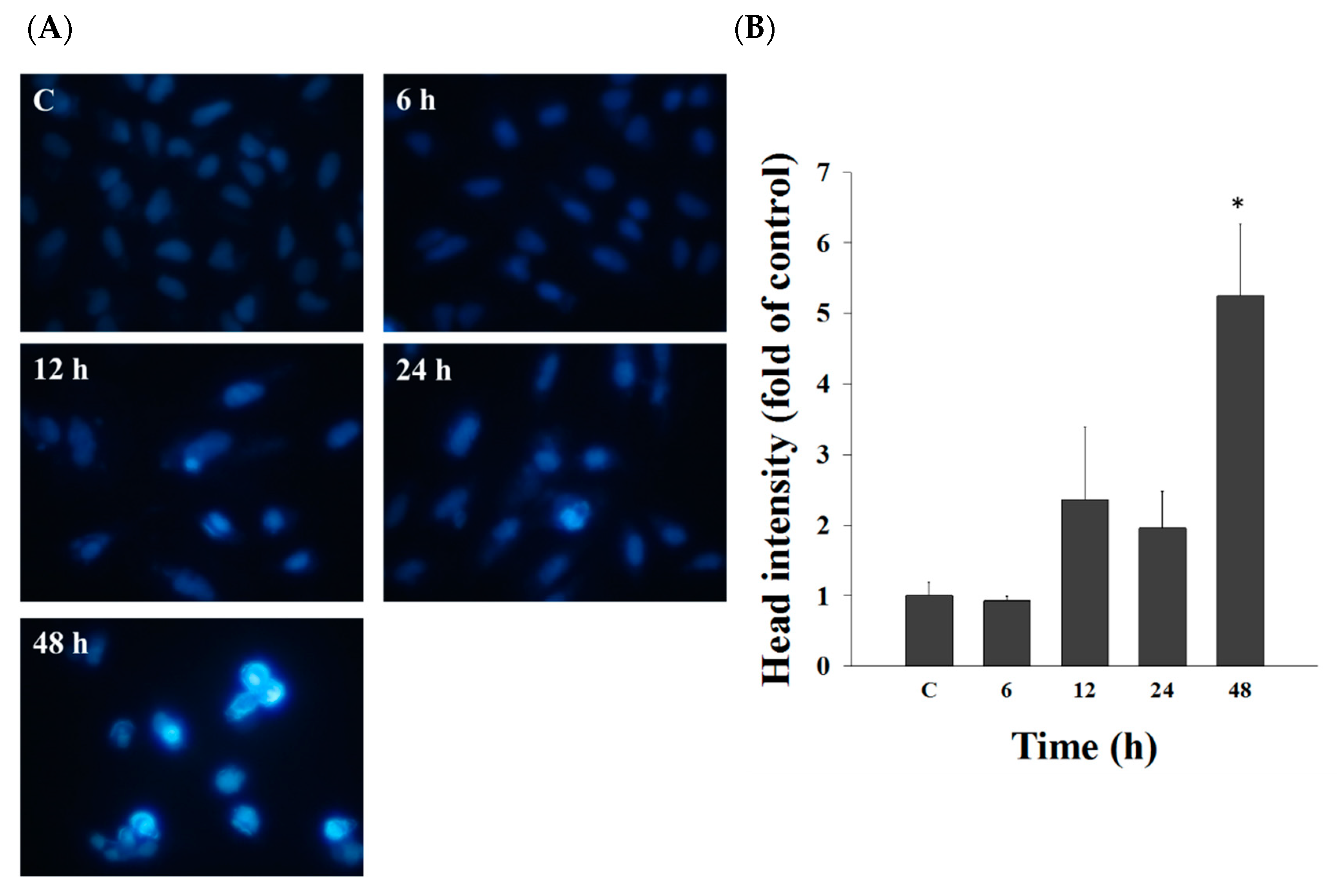
2.2. Casticin Induced Chromatin Condensation in A549 Cells
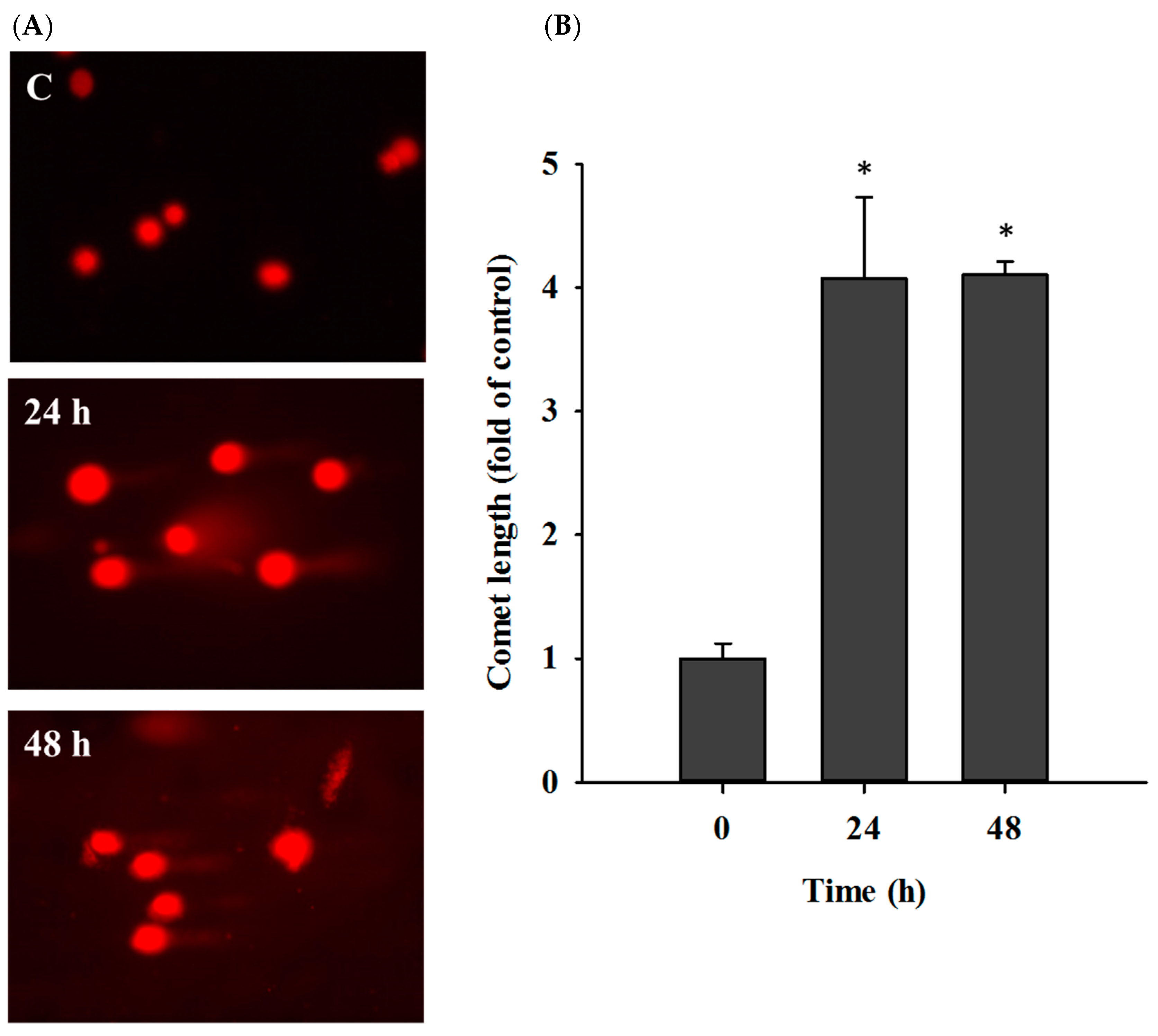
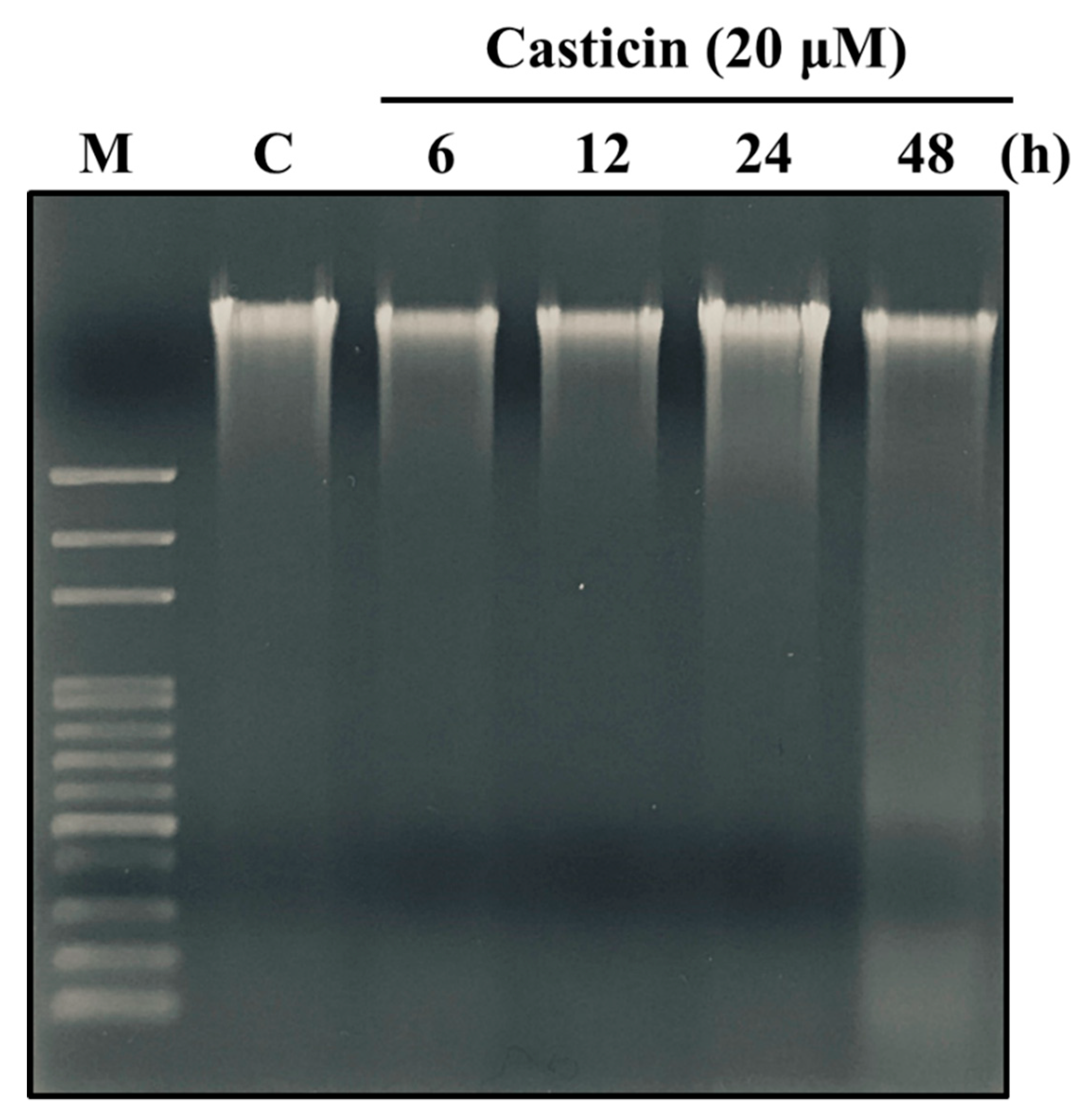
2.3. Casticin Induced DNA Damage in A549 Cells
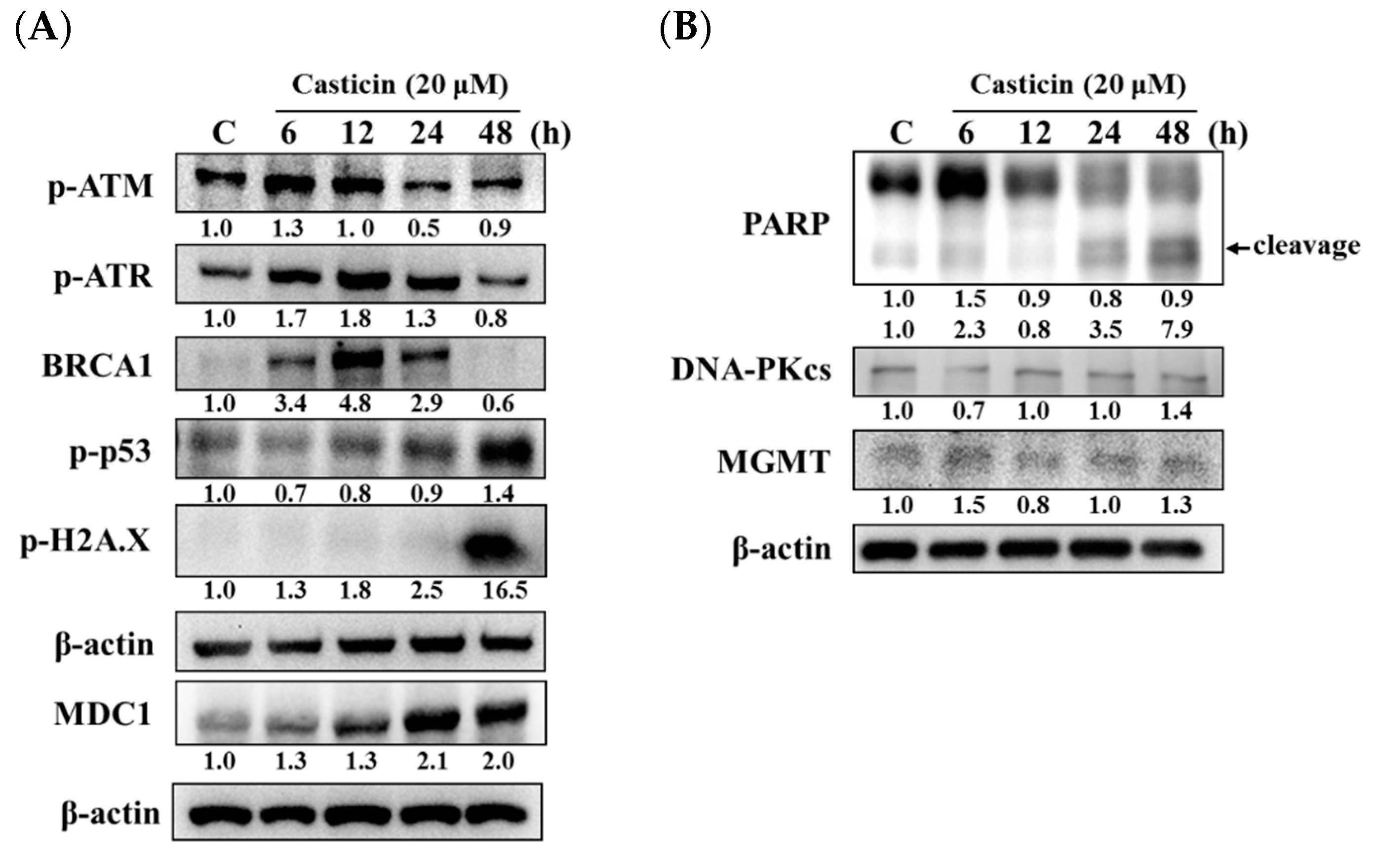
2.4. Casticin Affected the Levels of DNA Damage-Associated Proteins in A549 Cells
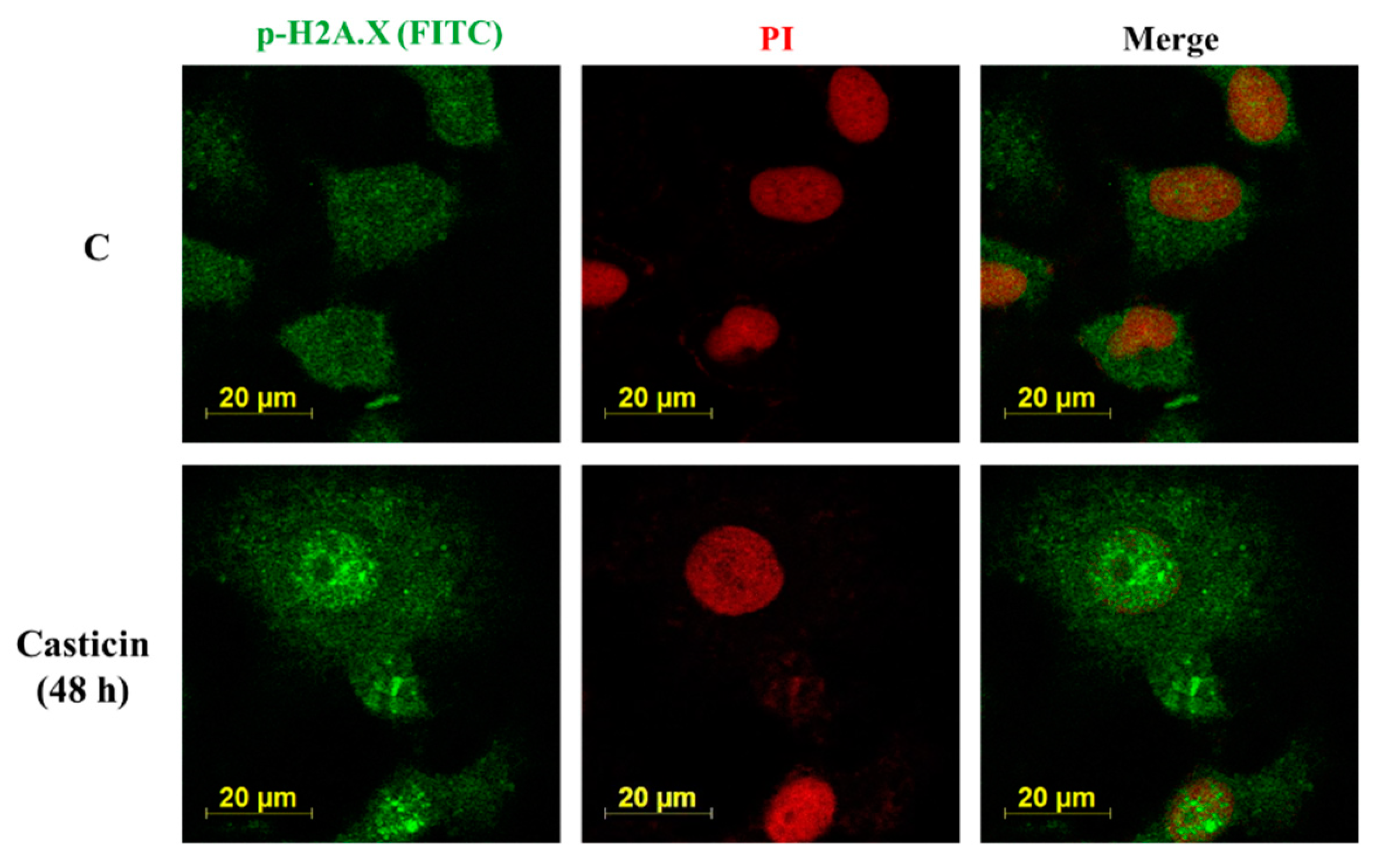
2.5. Casticin Affected the Expression and Translocation of p-H2AX on A549 Cells
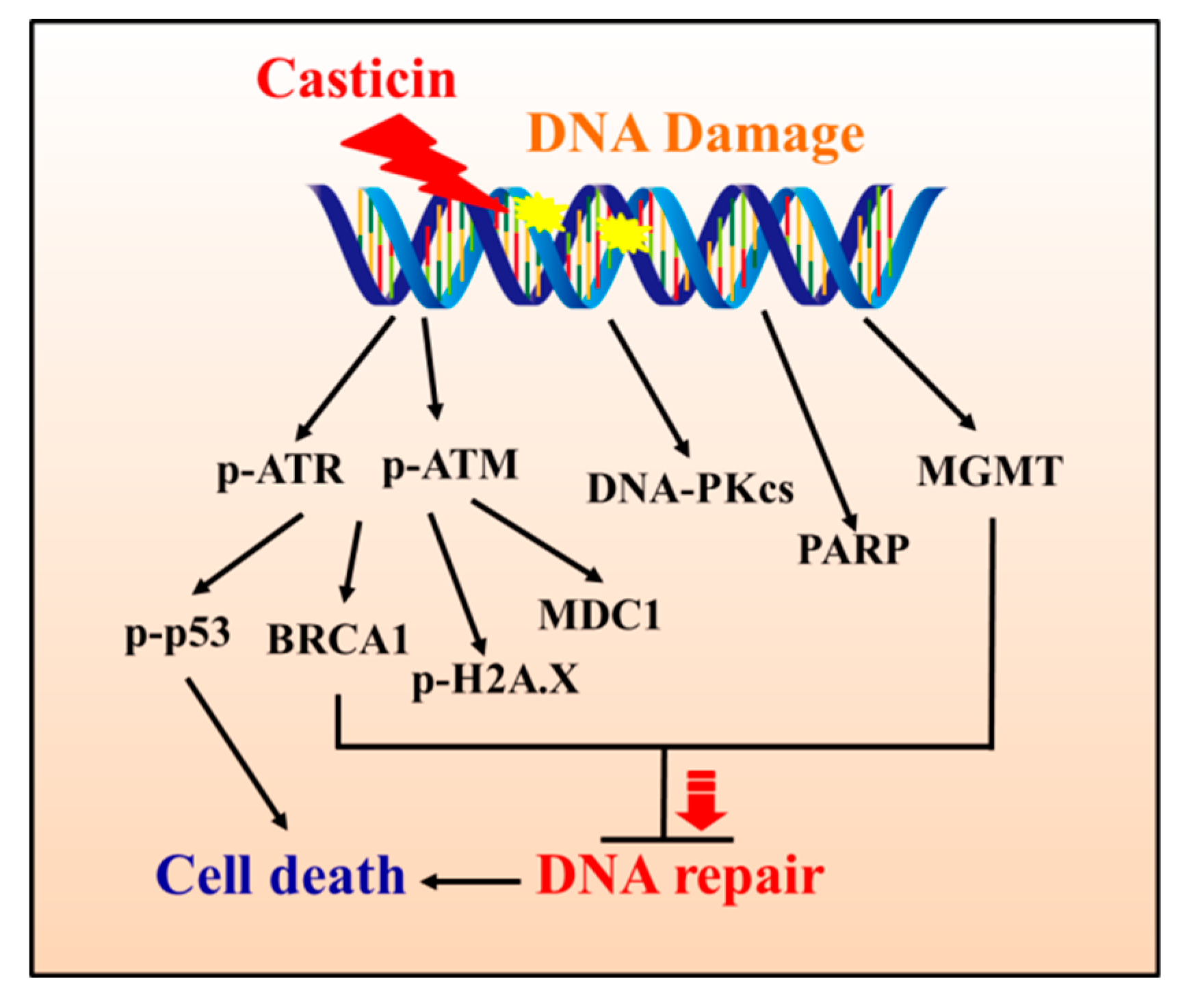
3. Discussion
4. Material and methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Measurements of Total Cell Viability
4.4. DAPI Staining
4.5. Comet Assay
4.6. DNA Gel Electrophoresis
4.7. Western Blotting
4.8. Immunofluorescence Assay
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, M.; Zhang, J.; Zhang, J.; Miao, Q.; Yao, L.; Zhang, J. Curcumin promotes apoptosis by activating the p53-miR-192-5p/215-XIAP pathway in non-small cell lung cancer. Cancer Lett. 2015, 357, 196–205. [Google Scholar] [CrossRef]
- Roointan, A.; Sharifi-Rad, M.; Badrzadeh, F.; Sharifi-Rad, J. A comparison between PLGA-PEG and NIPAAm-MAA nanocarriers in curcumin delivery for hTERT silencing in lung cancer cell line. Cell. Mol. Boil. 2016, 62, 51–56. [Google Scholar]
- Werner, M.E.; Cummings, N.D.; Sethi, M.; Wang, E.C.; Sukumar, R.; Moore, M.T.; Wang, A.Z. Preclinical evaluation of Genexol-PM, a nanoparticle formulation of paclitaxel, as a novel radiosensitizer for the treatment of non-small cell lung cancer. Int. J. Radiat. Oncol. 2013, 86, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Timmerman, R.; Paulus, R.; Galvin, J.; Michalski, J.; Straube, W.; Bradley, J.; Fakiris, A.; Bezjak, A.; Videtic, G.; Johnstone, D.; et al. Stereotactic body radiation therapy for inoperable early stage lung cancer. JAMA 2010, 303, 1070–1076. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.Y.; Senan, S.; Paul, M.A.; Mehran, R.J.; Louie, A.V.; Balter, P.; Groen, H.J.; McRae, S.E.; Widder, J.; Feng, L.; et al. Stereotactic ablative radiotherapy versus lobectomy for operable stage I non-small-cell lung cancer: A pooled analysis of two randomised trials. Lancet Oncol. 2015, 16, 630–637. [Google Scholar] [CrossRef] [Green Version]
- Roointan, A.; Ahmad Mir, T.; Ibrahim Wani, S.; Mati Ur, R.; Hussain, K.K.; Ahmed, B.; Abrahim, S.; Savardashtaki, A.; Gandomani, G.; Gandomani, M.; et al. Early detection of lung cancer biomarkers through biosensor technology: A review. J. Pharm. Biomed. Anal. 2019, 164, 93–103. [Google Scholar] [CrossRef]
- 2017 Ministry of Health and Welfare Report; Ministry of Health and Welfare: Taipei, Taiwan, 2018.
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, M.; Nambiar, M.; Sharma, S.; Karki, S.S.; Goldsmith, G.; Hegde, M.; Kumar, S.; Pandey, M.; Singh, R.K.; Ray, P.; et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell 2012, 151, 1474–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delia, D.; Mizutani, S. The DNA damage response pathway in normal hematopoiesis and malignancies. Int J. Hematol. 2017, 106, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesnow, S.; Argus, M.; Bergman, H.; Chu, K.; Frith, C.; Helmes, T.; McGaughy, R.; Ray, V.; Slaga, T.J.; Tennant, R.; et al. Chemical carcinogens. A review and analysis of the literature of selected chemicals and the establishment of the Gene-Tox Carcinogen Data Base. A report of the U.S. Environmental Protection Agency Gene-Tox Program. Mutat. Res. 1987, 185, 1–195. [Google Scholar] [CrossRef]
- Barnes, J.L.; Zubair, M.; John, K.; Poirier, M.C.; Martin, F.L. Carcinogens and DNA damage. Biochem. Soc. Trans. 2018, 46, 1213–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasul, A.; Zhao, B.J.; Liu, J.; Liu, B.; Sun, J.X.; Li, J.; Li, X.M. Molecular mechanisms of casticin action: An update on its antitumor functions. Asian Pac. J. Cancer Prev. APJCP 2014, 15, 9049–9058. [Google Scholar] [CrossRef] [Green Version]
- Mesaik, M.A.; Murad, S.; Khan, K.M.; Tareen, R.B.; Ahmed, A.; Choudhary, M.I. Isolation and immunomodulatory properties of a flavonoid, casticin from Vitex agnus-castus. Phytother. Res. PTR 2009, 23, 1516–1520. [Google Scholar] [CrossRef]
- Ye, Q.; Zhang, Q.Y.; Zheng, C.J.; Wang, Y.; Qin, L.P. Casticin, a flavonoid isolated from Vitex rotundifolia, inhibits prolactin release in vivo and in vitro. Acta Pharmacol. Sin. 2010, 31, 1564–1568. [Google Scholar] [CrossRef] [Green Version]
- de Sampaio e Spohr, T.C.; Stipursky, J.; Sasaki, A.C.; Barbosa, P.R.; Martins, V.; Benjamim, C.F.; Roque, N.F.; Costa, S.L.; Gomes, F.C. Effects of the flavonoid casticin from Brazilian Croton betulaster in cerebral cortical progenitors in vitro: Direct and indirect action through astrocytes. J. Neurosci. Res. 2010, 88, 530–541. [Google Scholar] [CrossRef]
- Kobayakawa, J.; Sato-Nishimori, F.; Moriyasu, M.; Matsukawa, Y. G2-M arrest and antimitotic activity mediated by casticin, a flavonoid isolated from Viticis Fructus (Vitex rotundifolia Linne fil.). Cancer Lett. 2004, 208, 59–64. [Google Scholar] [CrossRef]
- Tang, S.Y.; Zhong, M.Z.; Yuan, G.J.; Hou, S.P.; Yin, L.L.; Jiang, H.; Yu, Z. Casticin, a flavonoid, potentiates TRAIL-induced apoptosis through modulation of anti-apoptotic proteins and death receptor 5 in colon cancer cells. Oncol. Rep. 2013, 29, 474–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, D.J.; Ahn, H.S.; Chung, H.S.; Lee, H.; Kim, Y.; Lee, J.Y.; Kim, D.G.; Hong, M.; Shin, M.; Bae, H. Inhibitory effects of casticin on migration of eosinophil and expression of chemokines and adhesion molecules in A549 lung epithelial cells via NF-kappaB inactivation. J. Ethnopharmacol. 2011, 136, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zeng, L.; Zhang, T.; Liu, J.; Wang, W. Casticin inhibits lipopolysaccharide-induced acute lung injury in mice. Eur. J. Pharmacol. 2016, 789, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jung, K.H.; Lee, H.; Park, S.; Choi, W.; Bae, H. Casticin, an active compound isolated from Vitex Fructus, ameliorates the cigarette smoke-induced acute lung inflammatory response in a murine model. Int. Immunopharmacol. 2015, 28, 1097–1101. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, C.; Um, J.Y.; Sethi, G.; Ahn, K.S. Casticin-Induced Inhibition of Cell Growth and Survival Are Mediated through the Dual Modulation of Akt/mTOR Signaling Cascade. Cancers 2019, 11, 254. [Google Scholar] [CrossRef] [Green Version]
- Shih, Y.L.; Chou, J.; Yeh, M.Y.; Chou, H.M.; Chou, H.C.; Lu, H.F.; Shang, H.S.; Chueh, F.S.; Chu, Y.L.; Hsueh, S.C.; et al. Casticin induces DNA damage and inhibits DNA repair-associated protein expression in B16F10 mouse melanoma cancer cells. Oncol. Rep. 2016, 36, 2094–2100. [Google Scholar] [CrossRef]
- McCloskey, D.E.; Kaufmann, S.H.; Prestigiacomo, L.J.; Davidson, N.E. Paclitaxel induces programmed cell death in MDA-MB-468 human breast cancer cells. Clin. Cancer Res. 1996, 2, 847–854. [Google Scholar]
- Hu, L.; Hofmann, J.; Lu, Y.; Mills, G.B.; Jaffe, R.B. Inhibition of phosphatidylinositol 3′-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res. 2002, 62, 1087–1092. [Google Scholar]
- Hentze, H.; Latta, M.; Kunstle, G.; Dhakshinamoorthy, S.; Ng, P.Y.; Porter, A.G.; Wendel, A. Topoisomerase inhibitor camptothecin sensitizes mouse hepatocytes in vitro and in vivo to TNF-mediated apoptosis. Hepatology 2004, 39, 1311–1320. [Google Scholar] [CrossRef]
- Zhou, Y.; Peng, Y.; Mao, Q.Q.; Li, X.; Chen, M.W.; Su, J.; Tian, L.; Mao, N.Q.; Long, L.Z.; Quan, M.F.; et al. Casticin induces caspase-mediated apoptosis via activation of mitochondrial pathway and upregulation of DR5 in human lung cancer cells. Asian Pac. J. Trop. Med. 2013, 6, 372–378. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Cao, X.; Liu, Z.; Guo, H.; Ren, K.; Quan, M.; Zhou, Y.; Xiang, H.; Cao, J. Casticin suppresses self-renewal and invasion of lung cancer stem-like cells from A549 cells through down-regulation of pAkt. Acta Biochim. Biophys. Sin. 2014, 46, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Q.; Cao, X.; Cao, J.; Yang, X.; Zeng, W. Casticin suppresses the carcinogenesis of small cell lung cancer H446 cells through activation of AMPK/FoxO3a signaling. Oncol. Rep. 2018, 40, 1401–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.-Y.; Zhong, L.-J.; Xie, J.-M.; Wang, F.; Zhang, Y.-H. A New Taraxastane-Type Triterpene from Vitex trifolia var. simplicifolia. Helv. Chim. Acta 2013, 96, 2040–2045. [Google Scholar] [CrossRef]
- Mu, Y.; Hao, W.; Li, S. Casticin protects against IL-1beta-induced inflammation in human osteoarthritis chondrocytes. Eur. J. Pharmacol. 2019, 842, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Q.; Zhao, L.; Wang, Y.; Xue, L.; Han, T.; Zheng, C.; Qin, L. Quantitative determination and pharmacokinetic study of casticin in rat plasma by liquid chromatography-mass spectrometry. J. Pharm. Biomed. Anal. 2012, 61, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.C.; Peng, S.F.; Lai, K.C.; Liao, C.L.; Huang, Y.P.; Lin, C.C.; Lin, M.L.; Liu, K.C.; Tsai, C.C.; Ma, Y.S.; et al. Genistein induces apoptosis in vitro and has antitumor activity against human leukemia HL-60 cancer cell xenograft growth in vivo. Environ. Toxicol. 2019, 34, 443–456. [Google Scholar] [CrossRef]
- Su, E.Y.; Chu, Y.L.; Chueh, F.S.; Ma, Y.S.; Peng, S.F.; Huang, W.W.; Liao, C.L.; Huang, A.C.; Chung, J.G. Bufalin Induces Apoptotic Cell Death in Human Nasopharyngeal Carcinoma Cells through Mitochondrial ROS and TRAIL Pathways. Am. J. Chin. Med. 2019, 47, 237–257. [Google Scholar] [CrossRef]
- Yang, M.Y.; Wang, C.J.; Chen, N.F.; Ho, W.H.; Lu, F.J.; Tseng, T.H. Luteolin enhances paclitaxel-induced apoptosis in human breast cancer MDA-MB-231 cells by blocking STAT3. Chem. Biol. Interact. 2014, 213, 60–68. [Google Scholar] [CrossRef]
- Jakubowska, J.; Stasiak, M.; Szulawska, A.; Bednarek, A.; Czyz, M. Combined effects of doxorubicin and STI571 on growth, differentiation and apoptosis of CML cell line K562. Acta Biochim. Pol. 2007, 54, 839–846. [Google Scholar] [CrossRef] [Green Version]
- Soria, G.; Polo, S.E.; Almouzni, G. Prime, repair, restore: The active role of chromatin in the DNA damage response. Mol. Cell 2012, 46, 722–734. [Google Scholar] [CrossRef] [Green Version]
- Papamichos-Chronakis, M.; Peterson, C.L. Chromatin and the genome integrity network. Nat. Rev. Genet. 2013, 14, 62–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marnett, L.J.; Plastaras, J.P. Endogenous DNA damage and mutation. Trends Genet. 2001, 17, 214–221. [Google Scholar] [CrossRef]
- Wu, L.Y.; Lu, H.F.; Chou, Y.C.; Shih, Y.L.; Bau, D.T.; Chen, J.C.; Hsu, S.C.; Chung, J.G. Kaempferol induces DNA damage and inhibits DNA repair associated protein expressions in human promyelocytic leukemia HL-60 cells. Am. J. Chin. Med. 2015, 43, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, Y.; Tian, L.; Sheng, X.F.; Liu, F.; Cao, J.G. Casticin-induced apoptosis involves death receptor 5 upregulation in hepatocellular carcinoma cells. World J. Gastroenterol. 2011, 17, 4298–4307. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Cheng, Q.; Chen, J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 2010, 9, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.M.; Metcalfe, J.A.; Thick, J.; Mak, Y.F. Leukemia and lymphoma in ataxia telangiectasia. Blood 1996, 87, 423–438. [Google Scholar] [CrossRef]
- Barlow, C.; Hirotsune, S.; Paylor, R.; Liyanage, M.; Eckhaus, M.; Collins, F.; Shiloh, Y.; Crawley, J.N.; Ried, T.; Tagle, D.; et al. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell 1996, 86, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Ashley, T.; Brainerd, E.E.; Bronson, R.T.; Meyn, M.S.; Baltimore, D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996, 10, 2411–2422. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, V.J.; Rouleau, M.; Poirier, G.G. PARP-1, a determinant of cell survival in response to DNA damage. Exp. Hematol. 2003, 31, 446–454. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps317. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, S.; Mailand, N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair 2010, 9, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basseres, D.S.; Ebbs, A.; Cogswell, P.C.; Baldwin, A.S. IKK is a therapeutic target in KRAS-Induced lung cancer with disrupted p53 activity. Genes Cancer 2014, 5, 41–55. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, T.; Roth, J.A. Induction of apoptosis in human lung cancer cells after wild-type p53 activation by methoxyestradiol. Oncogene 1997, 14, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Melnikova, V.O.; Bolshakov, S.V.; Walker, C.; Ananthaswamy, H.N. Genomic alterations in spontaneous and carcinogen-induced murine melanoma cell lines. Oncogene 2004, 23, 2347–2356. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef]
- Queiroz, E.A.; Puukila, S.; Eichler, R.; Sampaio, S.C.; Forsyth, H.L.; Lees, S.J.; Barbosa, A.M.; Dekker, R.F.; Fortes, Z.B.; Khaper, N. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS ONE 2014, 9, e98207. [Google Scholar] [CrossRef]
- He, L.; Yang, X.; Cao, X.; Liu, F.; Quan, M.; Cao, J. Casticin induces growth suppression and cell cycle arrest through activation of FOXO3a in hepatocellular carcinoma. Oncol. Rep. 2013, 29, 103–108. [Google Scholar] [CrossRef]
- Nakamura, N.; Ramaswamy, S.; Vazquez, F.; Signoretti, S.; Loda, M.; Sellers, W.R. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol. Cell. Biol. 2000, 20, 8969–8982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Park, T.J.; Kim, J.Y.; Oh, S.P.; Kang, S.Y.; Kim, B.W.; Wang, H.J.; Song, K.Y.; Kim, H.C.; Lim, I.K. TIS21 negatively regulates hepatocarcinogenesis by disruption of cyclin B1-Forkhead box M1 regulation loop. Hepatology 2008, 47, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Liou, C.J.; Huang, W.C. Casticin inhibits interleukin-1beta-induced ICAM-1 and MUC5AC expression by blocking NF-kappaB, PI3K-Akt, and MAPK signaling in human lung epithelial cells. Oncotarget 2017, 8, 101175–101188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.N.; Wu, K.C.; Chung, W.S.; Zheng, L.C.; Juan, T.K.; Hsiao, Y.T.; Peng, S.F.; Yang, J.L.; Ma, Y.S.; Wu, R.S.; et al. Etomidate Suppresses Invasion and Migration of Human A549 Lung Adenocarcinoma Cells. Anticancer Res. 2019, 39, 215–223. [Google Scholar] [CrossRef]
- Lee, C.F.; Chiang, N.N.; Lu, Y.H.; Huang, Y.S.; Yang, J.S.; Tsai, S.C.; Lu, C.C.; Chen, F.A. Benzyl isothiocyanate (BITC) triggers mitochondria-mediated apoptotic machinery in human cisplatin-resistant oral cancer CAR cells. BioMedicine 2018, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Shih, Y.L.; Yeh, M.Y.; Liao, N.C.; Chung, H.Y.; Liu, K.L.; Lee, M.H.; Chou, P.Y.; Hou, H.Y.; Chou, J.S.; et al. Ursolic Acid Induces Apoptotic Cell Death Through AIF and Endo G Release Through a Mitochondria-dependent Pathway in NCI-H292 Human Lung Cancer Cells In Vitro. In Vivo 2019, 33, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Kuo, J.H.; Shih, T.Y.; Lin, J.P.; Lai, K.C.; Lin, M.L.; Yang, M.D.; Chung, J.G. Cantharidin induces DNA damage and inhibits DNA repair-associated protein expressions in TSGH8301 human bladder cancer cell. Anticancer Res. 2015, 35, 795–804. [Google Scholar]
- Lee, M.R.; Lin, C.; Lu, C.C.; Kuo, S.C.; Tsao, J.W.; Juan, Y.N.; Chiu, H.Y.; Lee, F.Y.; Yang, J.S.; Tsai, F.J. YC-1 induces G0/G1 phase arrest and mitochondria-dependent apoptosis in cisplatin-resistant human oral cancer CAR cells. BioMedicine 2017, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-P.; Hsu, C.-Y.; Fu, R.-H.; Huang, Y.-C.; Chen, S.-Y.; Lin, S.-Z.; Shyu, W.-C. Sambucus williamsii induced embryonic stem cells differentiated into neurons. BioMedicine 2015, 5, 1–5. [Google Scholar] [CrossRef]
Sample Availability: Samples of the casticin are not available from the authors. |







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Z.-Y.; Hsiao, Y.-T.; Huang, Y.-P.; Peng, S.-F.; Huang, W.-W.; Liu, K.-C.; Hsia, T.-C.; Way, T.-D.; Chung, J.-G. Casticin Induces DNA Damage and Affects DNA Repair Associated Protein Expression in Human Lung Cancer A549 Cells. Molecules 2020, 25, 341. https://doi.org/10.3390/molecules25020341
Cheng Z-Y, Hsiao Y-T, Huang Y-P, Peng S-F, Huang W-W, Liu K-C, Hsia T-C, Way T-D, Chung J-G. Casticin Induces DNA Damage and Affects DNA Repair Associated Protein Expression in Human Lung Cancer A549 Cells. Molecules. 2020; 25(2):341. https://doi.org/10.3390/molecules25020341
Chicago/Turabian StyleCheng, Zheng-Yu, Yung-Ting Hsiao, Yi-Ping Huang, Shu-Fen Peng, Wen-Wen Huang, Kuo-Ching Liu, Te-Chun Hsia, Tzong-Der Way, and Jing-Gung Chung. 2020. "Casticin Induces DNA Damage and Affects DNA Repair Associated Protein Expression in Human Lung Cancer A549 Cells" Molecules 25, no. 2: 341. https://doi.org/10.3390/molecules25020341