Gypenoside L Inhibits Proliferation of Liver and Esophageal Cancer Cells by Inducing Senescence

Abstract
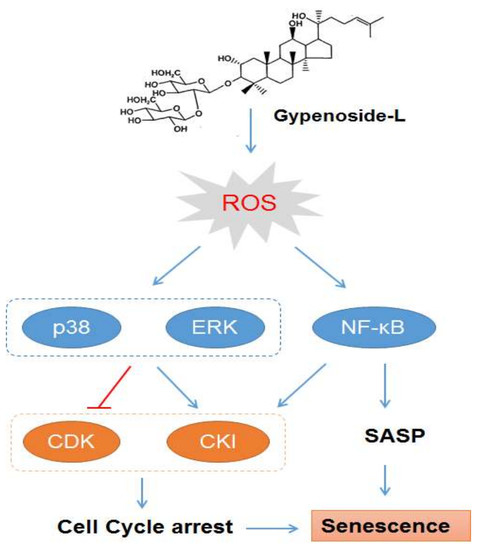
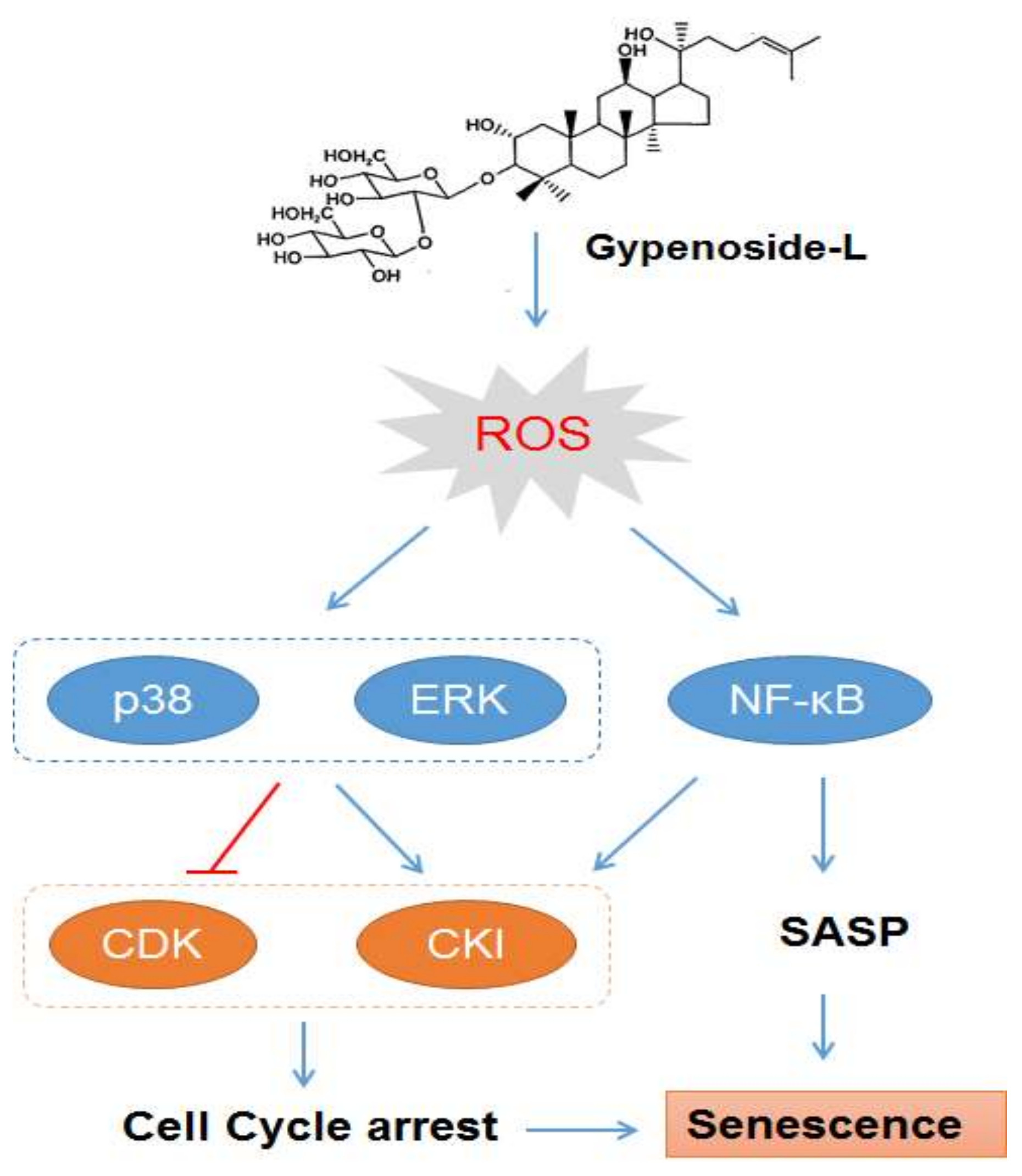
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
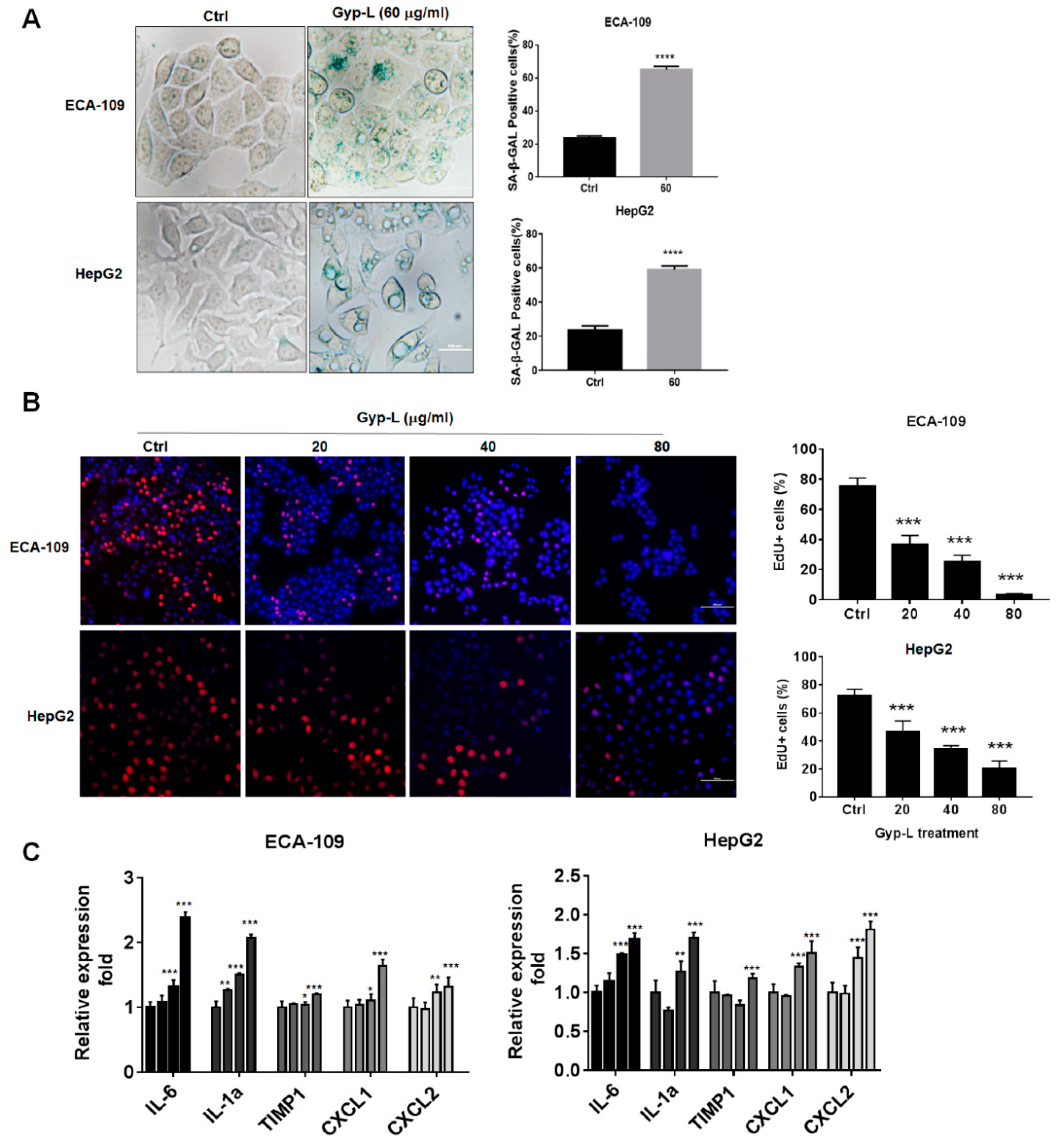
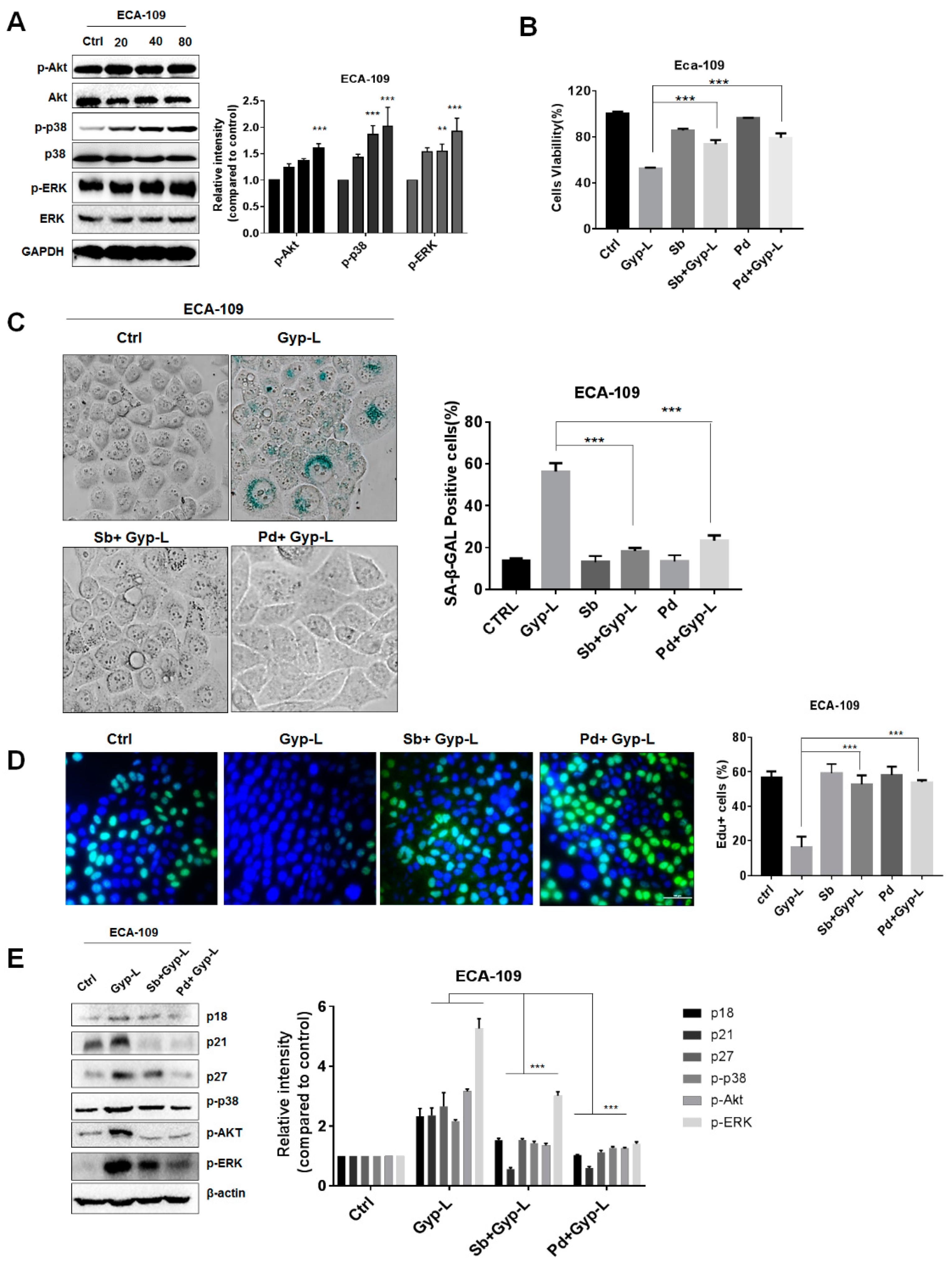
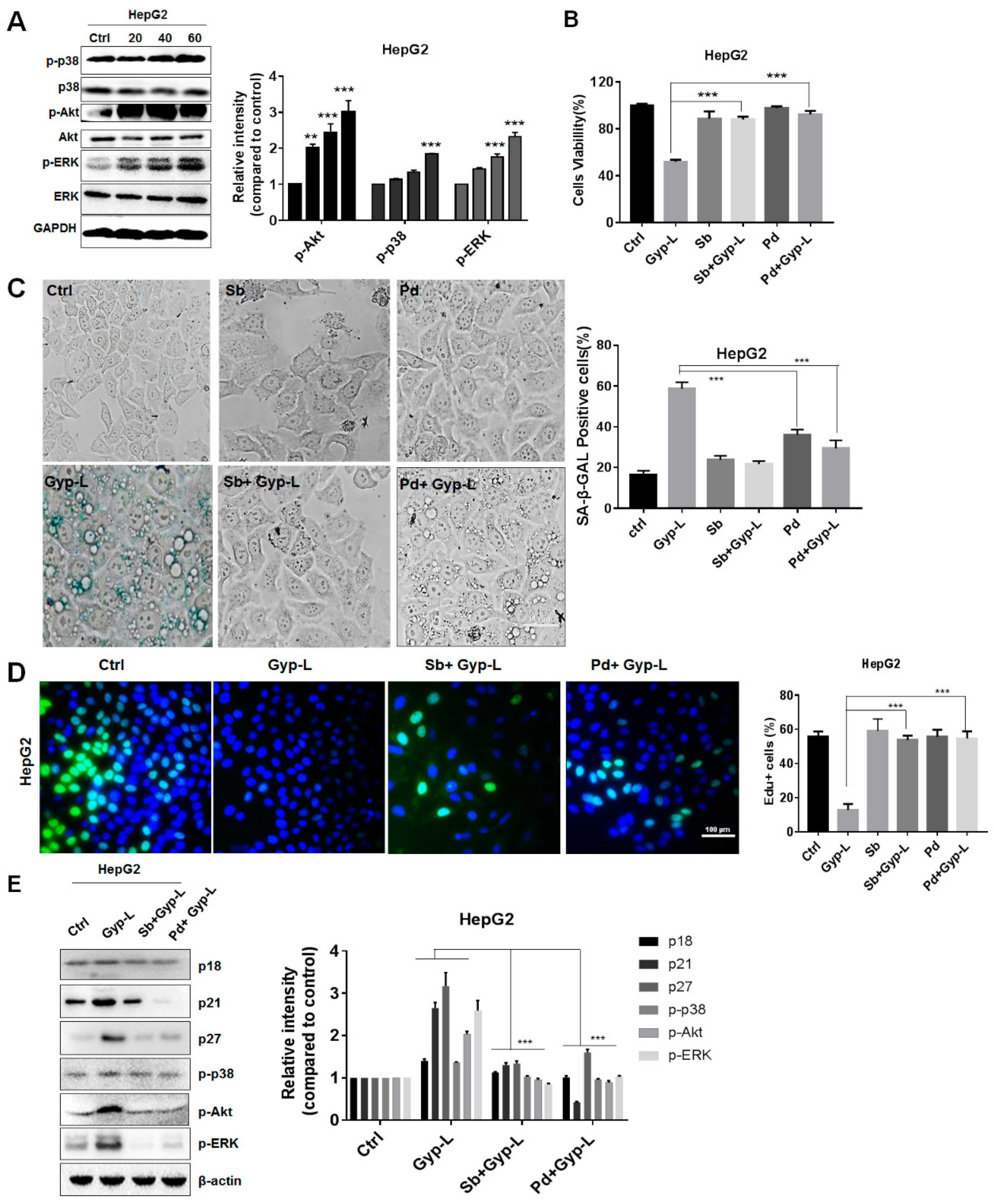
2.1. Gyp-L Induces Senescence in Cancer Cells
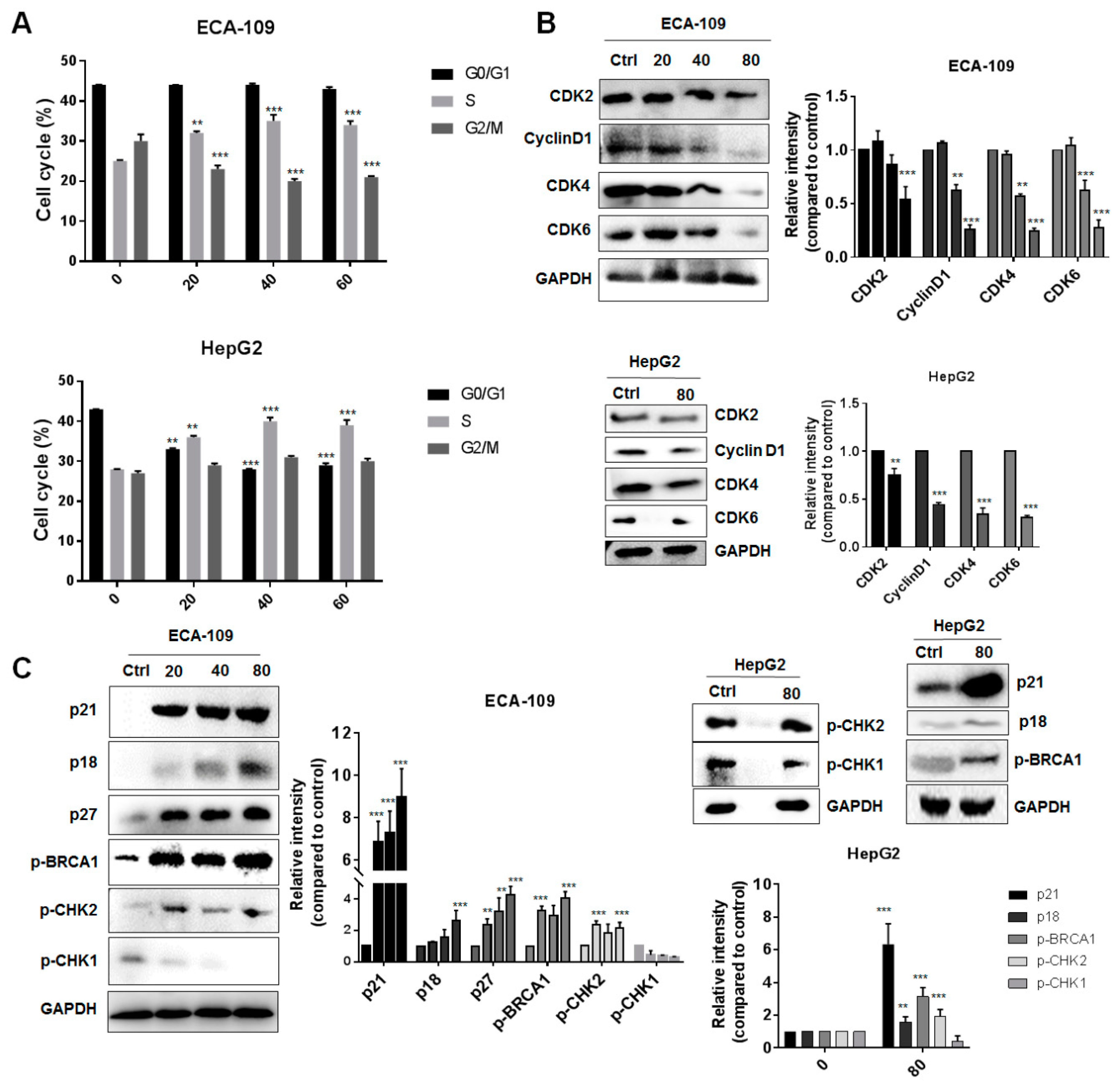
2.2. Gyp-L Causes Cell Cycle Arrest
2.3. Gyp-L Induces Senescence Via MAPK Signals
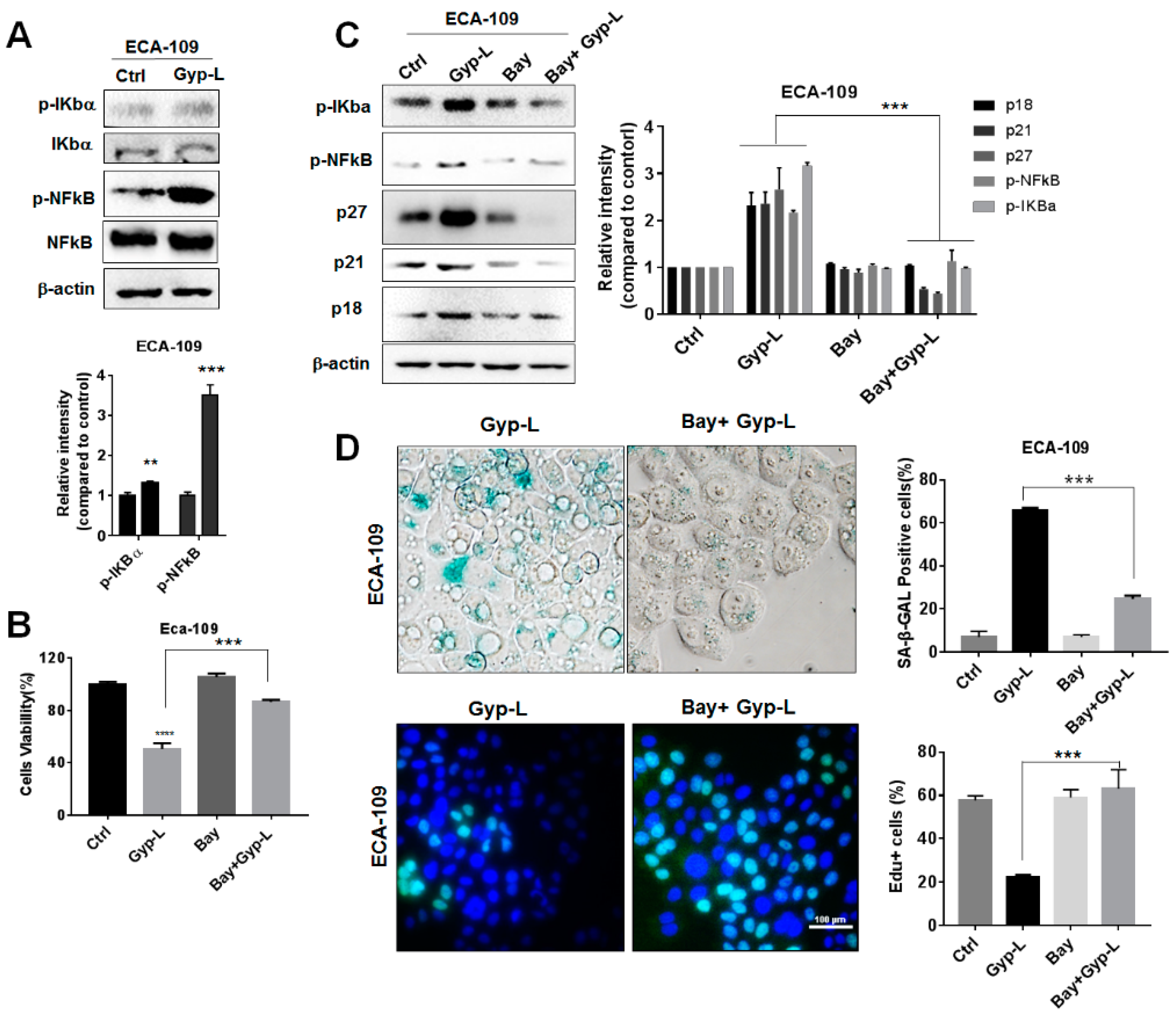
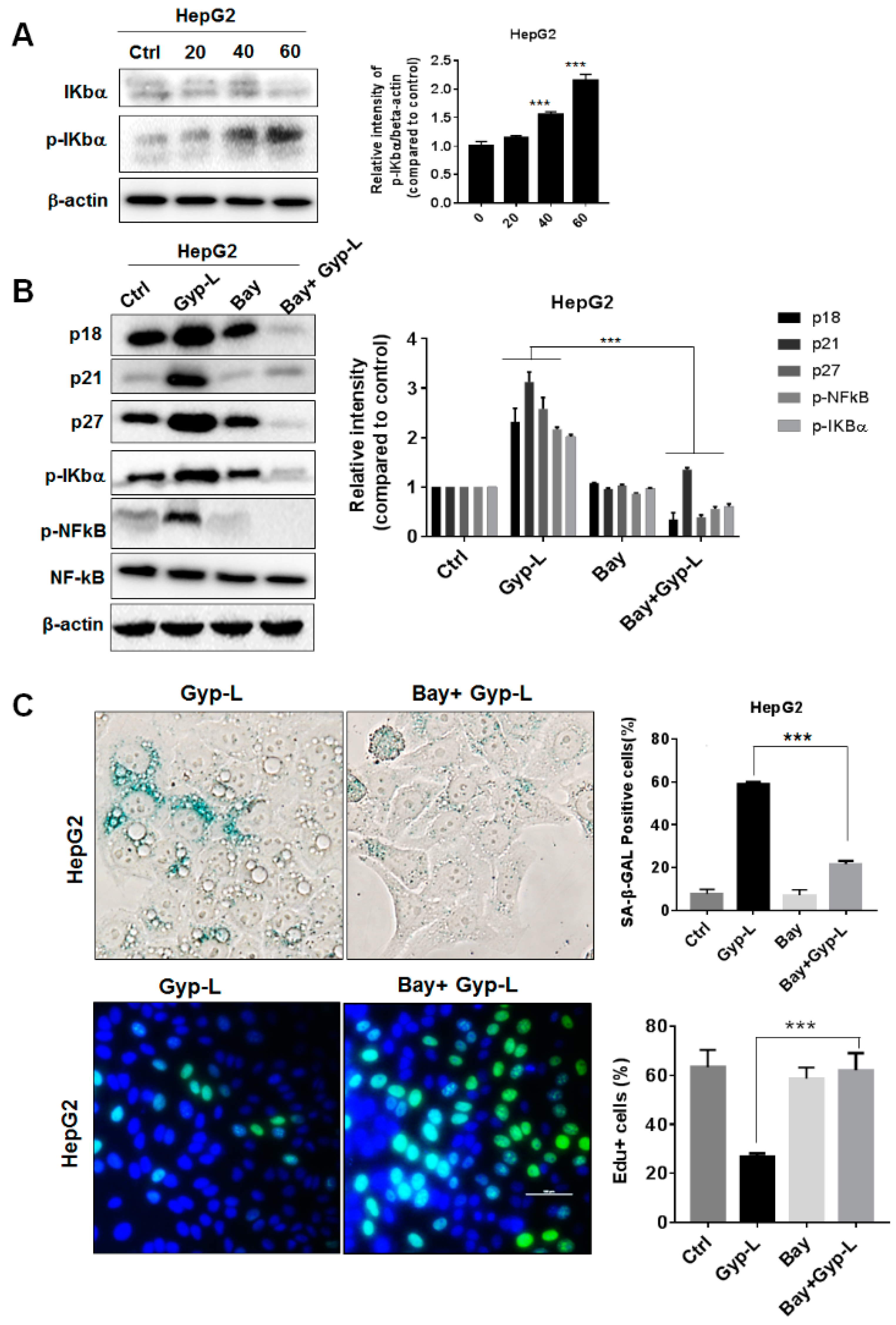
2.4. Gyp-L Induces Senescence Via NF-Κb Activation
2.5. Gyp-L Enhances the Sensitivity of Cancer Cells Toward Chemotherapy
2.6. Discussion
3. Materials and Methods
3.1. Cell Lines and Culture
3.2. Antibodies and Inhibitors
3.3. Western Blotting
3.4. Senescence-Associated β-Galactosidase (SA-β-gal) Staining
3.5. Cell Counting Kit-8 (CCK8) Assay
3.6. EdU Staining
3.7. FACS Analysis
3.8. Quantitative Real-Time PCR (qRT-PCR)
3.9. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brandsma, I.; Fleuren, E.D.G.; Williamson, C.T.; Lord, C.J. Directing the use of DDR kinase inhibitors in cancer treatment. Expert Opin. Investig. Drugs 2017, 26, 1341–1355. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Butera, G.; Pacchiana, R.; Donadelli, M. Autocrine mechanisms of cancer chemoresistance. Semin. Cell Dev. Biol. 2018, 78, 3–12. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W.; Roninson, I.B. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 2004, 23, 2919–2933. [Google Scholar] [CrossRef] [Green Version]
- Chandler, H.; Peters, G. Stressing the cell cycle in senescence and aging. Curr. Opin. Cell Biol. 2013, 25, 765–771. [Google Scholar] [CrossRef]
- Lasry, A.; Ben-Neriah, Y. Senescence-associated inflammatory responses: Aging and cancer perspectives. Trends Immunol. 2015, 36, 217–228. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Idelchik, M.D.P.S.; Melendez, J.A. Redox control of senescence and age-related disease. Redox Biol. 2017, 11, 91–102. [Google Scholar] [CrossRef]
- Zheng, K.; He, Z.; Kitazato, K.; Wang, Y. Selective autophagy regulates cell cycle in cancer therapy. Theranostics 2019, 9, 104–125. [Google Scholar] [CrossRef]
- Grossi, V.; Peserico, A.; Tezil, T.; Simone, C. p38α MAPK pathway: A key factor in colorectal cancer therapy and chemoresistance. World J. Gastroenterol. 2014, 20, 9744–9758. [Google Scholar] [CrossRef]
- Cragg, G.M.; Pezzuto, J.M. Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventive Agents. Med. Princ. Pract. 2016, 25 (Suppl. 2), 41–59. [Google Scholar] [CrossRef]
- Sanders, K.; Moran, Z.; Shi, Z.; Paul, R.; Greenlee, H. Natural Products for Cancer Prevention: Clinical Update 2016. Semin. Oncol. Nurs. 2016, 32, 215–240. [Google Scholar] [CrossRef]
- Shi, L.; Pi, Y.; Luo, C.; Zhang, C.; Tan, D.; Meng, X. In vitro inhibitory activities of six gypenosides on human liver cancer cell line HepG2 and possible role of HIF-1α pathway in them. Chem. Biol. Interact. 2015, 238, 48–54. [Google Scholar] [CrossRef]
- Liu, J.S.; Chiang, T.H.; Wang, J.S.; Lin, L.J.; Chao, W.C.; Inbaraj, B.S.; Lu, J.F.; Chen, B.H. Induction of p53-independent growth inhibition in lung carcinoma cell A549 by gypenosides. J. Cell Mol. Med. 2015, 19, 1697–1709. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Wang, X.; Niu, J.; Wang, Y.; Wang, P.; Liu, Q. Anticancer effect and the underlying mechanisms of gypenosides on human colorectal cancer SW-480 cells. PLoS One 2014, 9, e95609. [Google Scholar]
- Cheng, T.C.; Lu, J.F.; Wang, J.S.; Lin, L.J.; Kuo, H.I.; Chen, B.H. Antiproliferation effect and apoptosis mechanism of prostate cancer cell PC-3 by flavonoids and saponins prepared from Gynostemma pentaphyllum. J. Agric. Food Chem. 2011, 59, 11319–11329. [Google Scholar] [CrossRef]
- Xie, Z.H.; Liu, W.; Huang, H.; Slavin, M.; Zhao, Y.; Whent, M.; Blackford, J.; Lutterodt, H.; Zhou, H.P.; Chen, P.; et al. Chemical composition of five commercial Gynostemma pentaphyllum samples and their radical scavenging, antiproliferative, and anti-inflammatory properties. J. Agric. Food Chem. 2010, 58, 11243–11249. [Google Scholar] [CrossRef]
- Zheng, K.; Liao, C.; Li, Y.; Fan, X.; Fan, L.; Xu, H.; Kang, Q.; Zeng, Y.; Wu, X.; Wu, H.; et al. Gypenoside l, isolated from Gynostemma Pentaphyllum, induces cytoplasmic vacuolation death in hepatocellular carcinoma cells through reactive-oxygen-species-mediated unfolded protein response. J. Agric. Food Chem. 2016, 64, 1702–1711. [Google Scholar] [CrossRef]
- Liao, C.; Zheng, K.; Li, Y.; Xu, H.; Kang, Q.; Fan, L.; Hu, X.; Jin, Z.; Zeng, Y.; Kong, X.; et al. Gypenoside L inhibits autophagic flux and induces cell death in human esophageal cancer cells through endoplasm reticulum stress-mediated Ca2+ release. Oncotarget 2016, 7, 47387–47402. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; Jiang, Y.; Liao, C.; Hu, X.; Li, Y.; Zeng, Y.; Zhang, J.; Wu, X.; Wu, H.; Liu, L.; et al. NOX2-mediated TFEB activation and vacuolization regulate lysosome-associated cell death induced by Gypenoside l, a saponin isolated from Gynostemma pentaphyllum. J. Agric. Food Chem. 2017, 65, 6625–6637. [Google Scholar] [CrossRef]
- Yuan, R.; Hou, Y.; Sun, W.; Yu, J.; Liu, X.; Niu, Y.; Lu, J.J.; Chen, X. Natural products to prevent drug resistance in cancer chemotherapy: A review. Ann. N. Y. Acad. Sci. 2017, 1401, 19–27. [Google Scholar] [CrossRef]
- Chang, U.M.; Li, C.H.; Lin, L.I.; Huang, C.P.; Kan, L.S.; Lin, S.B. Ganoderiol F, a ganoderma triterpene, induces senescence in hepatoma HepG2 cells. Life Sci. 2006, 79, 1129–1139. [Google Scholar] [CrossRef]
- Luo, H.; Yang, A.; Schulte, B.; Wargovich, M.; Wang, G. Resveratrol induces premature senescence in lung cancer cells via ROS-mediated DNA damage. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Alcántara-Flores, E.; Brechú-Franco, A.E.; García-López, P.; Rocha-Zavaleta, L.; López-Marure, R.; Martínez-Vázquez, M. Argentatin B inhibits proliferation of prostate and colon cancer cells by inducing cell senescence. Molecules 2015, 20, 21125–21137. [Google Scholar] [CrossRef]
- Roninson, I.B.; Broude, E.V.; Chang, B.D. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist. Updates 2001, 4, 303–313. [Google Scholar] [CrossRef]
- Xu, Y.; Li, N.; Xiang, R.; Sun, P. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends Biochem. Sci. 2014, 39, 268–276. [Google Scholar] [CrossRef] [Green Version]
- Osorio, F.G.; Soria-Valles, C.; Santiago-Fernández, O.; Freije, J.M.; López-Otín, C. NF-κB signaling as a driver of ageing. Int. Rev. Cell Mol. Biol. 2016, 326, 133–174. [Google Scholar]
- Zdanov, S.; Debacq-Chainiaux, F.; Remacle, J.; Toussaint, O. Identification of p38MAPK-dependent genes with changed transcript abundance in H2O2-induced premature senescence of IMR-90 hTERT human fibroblasts. FEBS Lett. 2006, 580, 6455–6463. [Google Scholar] [CrossRef]
- Colavitti, R.; Finkel, T. Reactive oxygen species as mediators of cellular senescence. IUBMB Life 2005, 57, 277–281. [Google Scholar] [CrossRef]
- Nicke, B.; Bastien, J.; Khanna, S.J.; Warne, P.H.; Cowling, V.; Cook, S.J.; Peters, G.; Delpuech, O.; Schulze, A.; Berns, K.; et al. Involvement of MINK, a Ste20 family kinase, in Ras oncogene-induced growth arrest in human ovarian surface epithelial cells. Mol. Cell. 2005, 20, 673–685. [Google Scholar] [CrossRef]
- Zheng, K.; Li, Y.; Wang, S.; Wang, X.; Liao, C.; Hu, X.; Fan, L.; Kang, Q.; Zeng, Y.; Wu, X.; et al. Inhibition of autophagosome-lysosome fusion by ginsenoside Ro via the ESR2-NCF1-ROS pathway sensitizes esophageal cancer cells to 5-fluorouracil-induced cell death via the CHEK1-mediated DNA damage checkpoint. Autophagy 2016, 12, 1593–1613. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.F.; Lin, Y.C.; Tsai, T.F.; Chen, H.E.; Chou, K.Y.; Hwang, T.I. Cisplatin induces protective autophagy through activation of BECN1 in human bladder cancer cells. Drug Des. Devel. Ther. 2017, 11, 1517–1533. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Wang, W. Knockdown of galectin-1 facilitated cisplatin sensitivity by inhibiting autophagy in neuroblastoma cells. Chem. Biol. Interact. 2019, 297, 50–56. [Google Scholar] [CrossRef]
- Jiang, Y.; Ji, F.; Liu, Y.; He, M.; Zhang, Z.; Yang, J.; Wang, N.; Zhong, C.; Jin, Q.; Ye, X.; et al. Cisplatin-induced autophagy protects breast cancer cells from apoptosis by regulating yes-associated protein. Oncol. Rep. 2017, 38, 3668–3676. [Google Scholar] [CrossRef]
Sample Availability: Sample of the compound Gypenoside L (Gyp-L) is available from the authors. |







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.; Hu, X.; Liao, C.; Xiao, H.; Zhu, Q.; Li, Y.; Liu, Z.; Tao, A.; He, Z.; Xu, C.; et al. Gypenoside L Inhibits Proliferation of Liver and Esophageal Cancer Cells by Inducing Senescence. Molecules 2019, 24, 1054. https://doi.org/10.3390/molecules24061054
Ma J, Hu X, Liao C, Xiao H, Zhu Q, Li Y, Liu Z, Tao A, He Z, Xu C, et al. Gypenoside L Inhibits Proliferation of Liver and Esophageal Cancer Cells by Inducing Senescence. Molecules. 2019; 24(6):1054. https://doi.org/10.3390/molecules24061054
Chicago/Turabian StyleMa, Jingxin, Xiaopeng Hu, Chenghui Liao, Haitao Xiao, Qinchang Zhu, Ying Li, Zhigang Liu, Anjin Tao, Zhendan He, Chenshu Xu, and et al. 2019. "Gypenoside L Inhibits Proliferation of Liver and Esophageal Cancer Cells by Inducing Senescence" Molecules 24, no. 6: 1054. https://doi.org/10.3390/molecules24061054