Emerging Roles of C-Myc in Cancer Stem Cell-Related Signaling and Resistance to Cancer Chemotherapy: A Potential Therapeutic Target Against Colorectal Cancer



Abstract
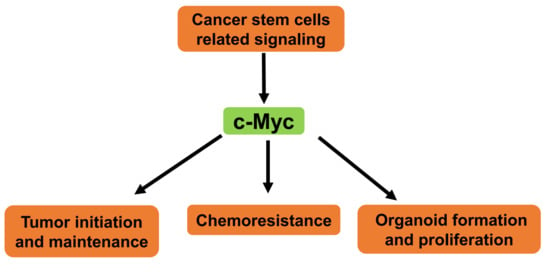
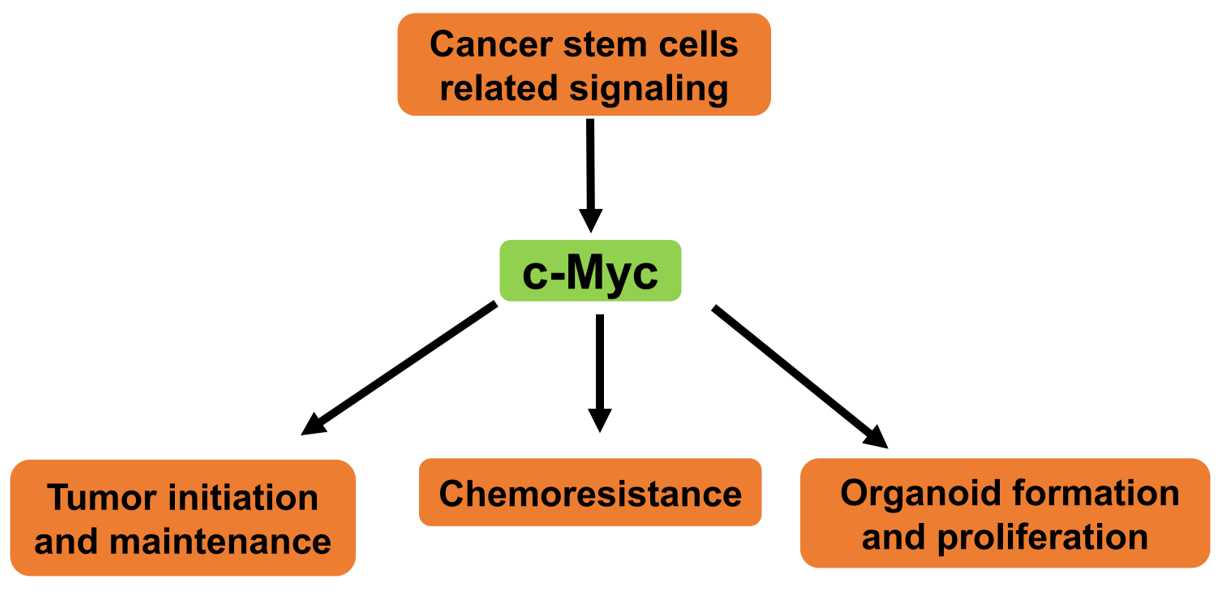
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
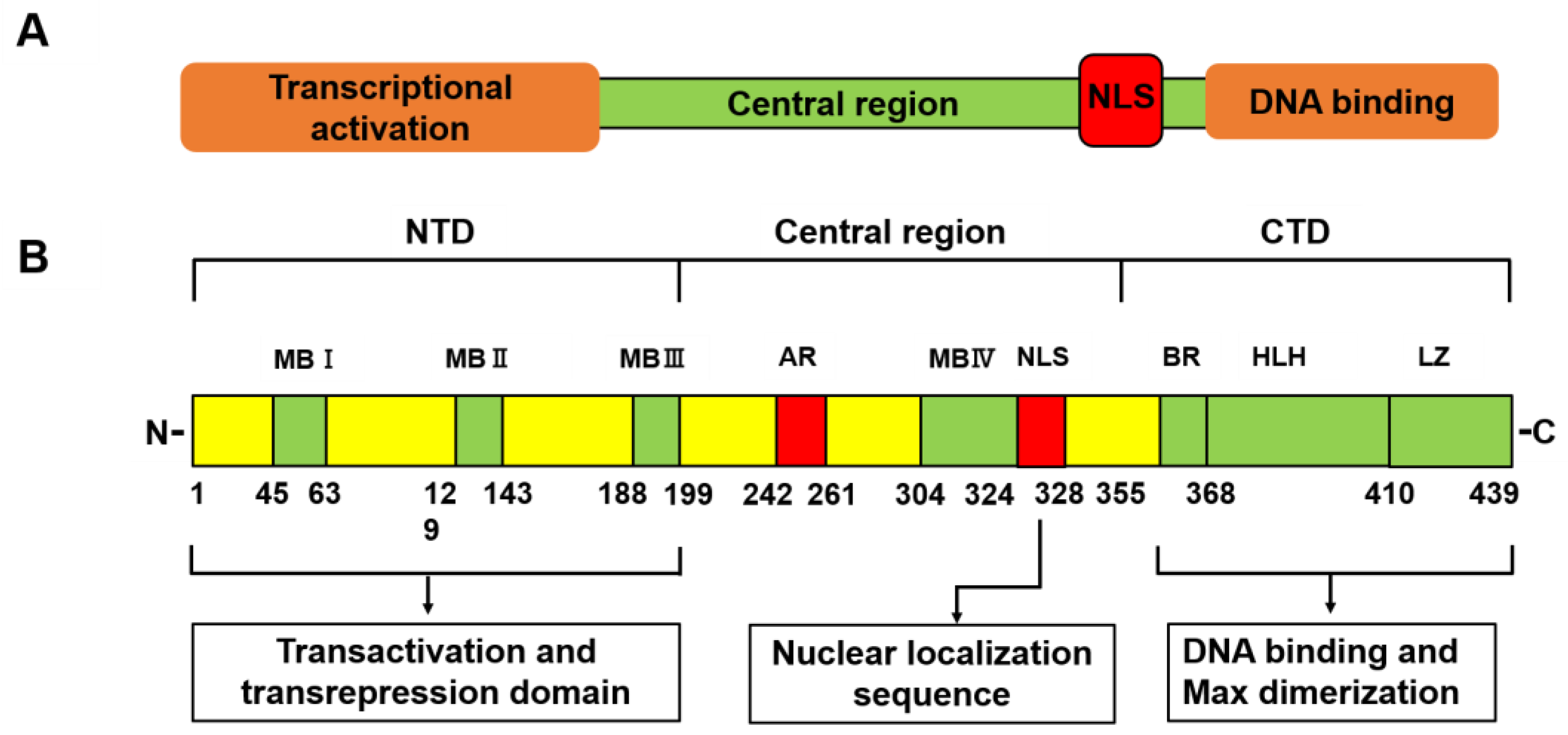
2. Structures of Myc Family Proteins
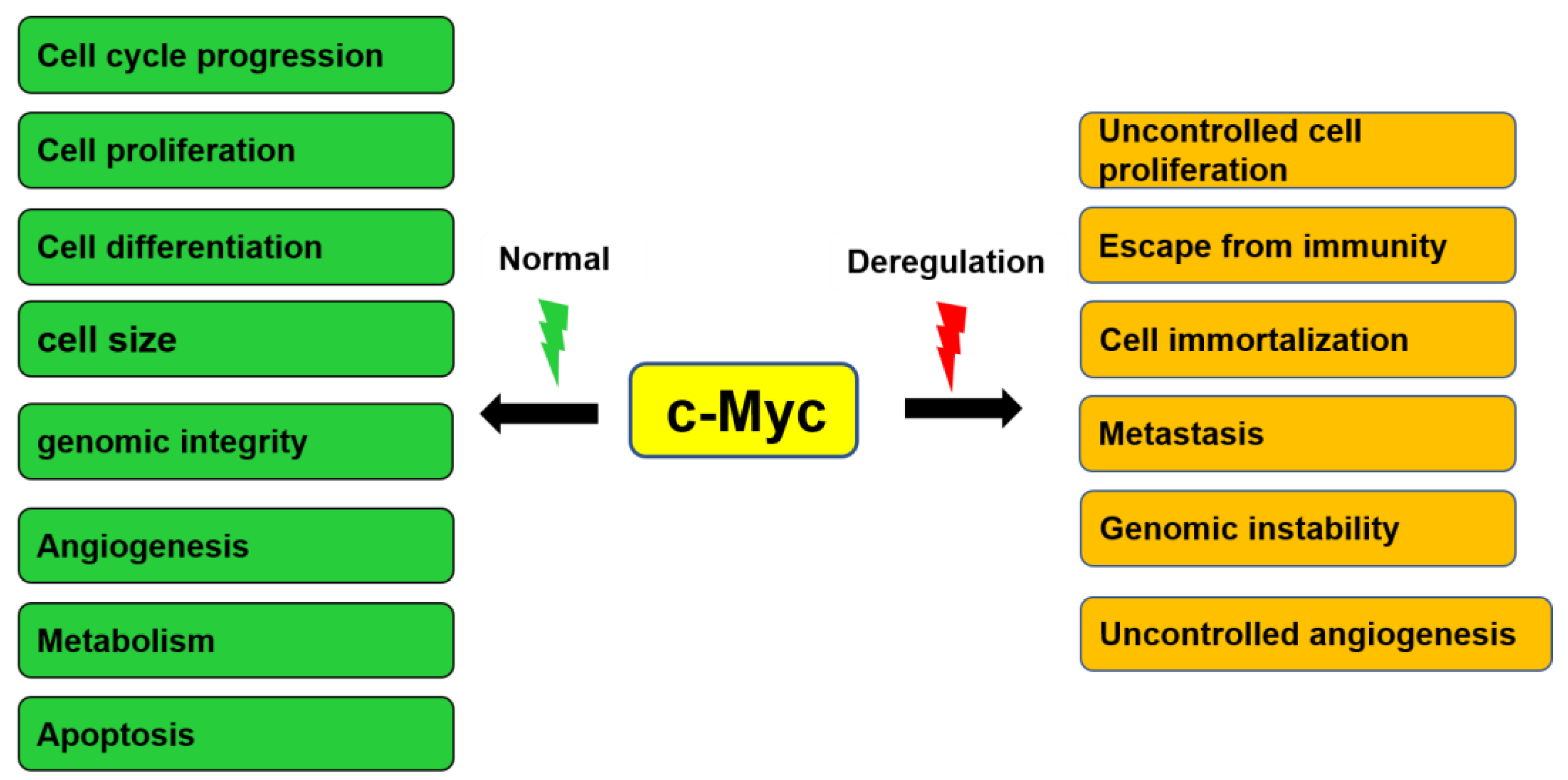
3. General Functions of c-Myc
4. Roles of c-Myc in Cancer
5. Roles of c-Myc in Colorectal Cancer
6. Roles of c-Myc in the Regulation of Cancer Stem Cell-Related Signaling
7. Roles of c-Myc in Resistance of Chemotherapy
8. Roles of c-Myc in Colorectal Cancer Organoids
9. Therapeutic Opportunities Targeting c-Myc
10. Conclusions
Funding
Conflicts of Interest
References
- Prendergast, G.C. Mechanisms of apoptosis by c-myc. Oncogene 1999, 18, 2967–2987. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The myc/max/mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef]
- Nasi, S.; Ciarapica, R.; Jucker, R.; Rosati, J.; Soucek, L. Making decisions through myc. Febs. Lett. 2001, 490, 153–162. [Google Scholar] [CrossRef]
- Albihn, A.; Johnsen, J.I.; Henriksson, M.A. Myc in oncogenesis and as a target for cancer therapies. Adv. Cancer Res. 2010, 107, 163–224. [Google Scholar] [PubMed]
- Dang, C.V. Myc, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Bretones, G.; Delgado, M.D.; Leon, J. Myc and cell cycle control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksson, M.; Luscher, B. Proteins of the myc network: Essential regulators of cell growth and differentiation. Adv. Cancer Res. 1996, 68, 109–182. [Google Scholar]
- Sheiness, D.; Fanshier, L.; Bishop, J.M. Identification of nucleotide sequences which may encode the oncogenic capacity of avian retrovirus mc29. J. Virol. 1978, 28, 600–610. [Google Scholar]
- Roussel, M.; Saule, S.; Lagrou, C.; Rommens, C.; Beug, H.; Graf, T.; Stehelin, D. Three new types of viral oncogene of cellular origin specific for haematopoietic cell transformation. Nature 1979, 281, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Nau, M.M.; Brooks, B.J.; Battey, J.; Sausville, E.; Gazdar, A.F.; Kirsch, I.R.; McBride, O.W.; Bertness, V.; Hollis, G.F.; Minna, J.D. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature 1985, 318, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Alitalo, K.; Klempnauer, K.H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [PubMed]
- Taub, R.; Kirsch, I.; Morton, C.; Lenoir, G.; Swan, D.; Tronick, S.; Aaronson, S.; Leder, P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7837–7841. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Harris, A.W.; Pinkert, C.A.; Corcoran, L.M.; Alexander, W.S.; Cory, S.; Palmiter, R.D.; Brinster, R.L. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985, 318, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- Dang, C.V. Myc on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic myc as a strategy for cancer treatment. Signal Transduct. Target. 2018, 3, 5. [Google Scholar] [CrossRef]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-myc protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef]
- Berg, T. Small-molecule modulators of c-myc/max and max/max interactions. Curr. Top. Microbiol. Immunol. 2011, 348, 139–149. [Google Scholar]
- Watt, R.; Nishikura, K.; Sorrentino, J.; ar-Rushdi, A.; Croce, C.M.; Rovera, G. The structure and nucleotide sequence of the 5’ end of the human c-myc oncogene. Proc. Natl. Acad. Sci. USA 1983, 80, 6307–6311. [Google Scholar] [CrossRef] [PubMed]
- Cashman, D.J.; Buscaglia, R.; Freyer, M.W.; Dettler, J.; Hurley, L.H.; Lewis, E.A. Molecular modeling and biophysical analysis of the c-myc nhe-iii1 silencer element. J. Mol. Model. 2008, 14, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Oster, S.K.; Ho, C.S.; Soucie, E.L.; Penn, L.Z. The myc oncogene: Marvelously complex. Adv. Cancer Res. 2002, 84, 81–154. [Google Scholar] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. Bet bromodomain inhibition as a therapeutic strategy to target c-myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Sakamuro, D.; Prendergast, G.C. New myc-interacting proteins: A second myc network emerges. Oncogene 1999, 18, 2942–2954. [Google Scholar] [CrossRef]
- Conzen, S.D.; Gottlob, K.; Kandel, E.S.; Khanduri, P.; Wagner, A.J.; Leary, M.; Hay, N. Induction of cell cycle progression and acceleration of apoptosis are two separable functions of c-myc: Transrepression correlates with acceleration of apoptosis. Mol. Cell. Biol. 2000, 20, 6008. [Google Scholar] [CrossRef] [PubMed]
- Pelengaris, S.; Khan, M.; Evan, G. C-myc: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.A.; Gallant, P. Control of growth and organ size in drosophila. BioEssays 2002, 24, 54–64. [Google Scholar] [CrossRef]
- Herbst, A.; Hemann, M.T.; Tworkowski, K.A.; Salghetti, S.E.; Lowe, S.W.; Tansey, W.P. A conserved element in myc that negatively regulates its proapoptotic activity. Embo Rep. 2005, 6, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Dolde, C.; Gillison, M.L.; Kato, G.J. Discrimination between related DNA sites by a single amino acid residue of myc-related basic-helix-loop-helix proteins. Proc. Natl. Acad. Sci. USA 1992, 89, 599. [Google Scholar] [CrossRef]
- Lüscher, B. Function and regulation of the transcription factors of the myc/max/mad network. Gene 2001, 277, 1–14. [Google Scholar] [CrossRef]
- Cowling, V.H.; Chandriani, S.; Whitfield, M.L.; Cole, M.D. A conserved Myc protein domain, MBIV, regulates DNA binding, apoptosis, transformation, and G2 arrest. Mol. Cell. Biol. 2006, 26, 4226. [Google Scholar] [CrossRef] [PubMed]
- Iritani, B.M.; Eisenman, R.N. C-myc enhances protein synthesis and cell size during b lymphocyte development. Proc. Natl. Acad. Sci. USA 1999, 96, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.Y.; Grove, L.; Datta, N.S.; Long, M.W.; Prochownik, E.V. C-myc overexpression and p53 loss cooperate to promote genomic instability. Oncogene 1999, 18, 1177–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudino, T.A.; McKay, C.; Pendeville-Samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. C-myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, A.; Deb-Basu, D.; Cherry, A.; Turner, S.; Ford, J.; Felsher, D.W. Defective double-strand DNA break repair and chromosomal translocations by myc overexpression. Proc. Natl. Acad. Sci. USA 2003, 100, 9974–9979. [Google Scholar] [CrossRef] [PubMed]
- Amati, B.; Alevizopoulos, K.; Vlach, J. Myc and the cell cycle. Front. Biosci. 1998, 3, d250–d268. [Google Scholar] [CrossRef]
- Cole, M.D.; Nikiforov, M.A. Transcriptional activation by the myc oncoprotein. In The myc/max/mad Transcription Factor Network; Eisenman, R.N., Ed.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 33–50. [Google Scholar]
- Zimmerman, K.A.; Yancopoulos, G.D.; Collum, R.G.; Smith, R.K.; Kohl, N.E.; Denis, K.A.; Nau, M.M.; Witte, O.N.; Toran-Allerand, D.; Gee, C.E.; et al. Differential expression of myc family genes during murine development. Nature 1986, 319, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.C.; Wims, M.; Spotts, G.D.; Hann, S.R.; Bradley, A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993, 7, 671–682. [Google Scholar] [CrossRef]
- Charron, J.; Malynn, B.A.; Fisher, P.; Stewart, V.; Jeannotte, L.; Goff, S.P.; Robertson, E.J.; Alt, F.W. Embryonic lethality in mice homozygous for a targeted disruption of the n-myc gene. Genes Dev. 1992, 6, 2248–2257. [Google Scholar] [CrossRef]
- Hatton, K.S.; Mahon, K.; Chin, L.; Chiu, F.C.; Lee, H.W.; Peng, D.; Morgenbesser, S.D.; Horner, J.; DePinho, R.A. Expression and activity of l-myc in normal mouse development. Mol. Cell. Biol. 1996, 16, 1794–1804. [Google Scholar] [CrossRef]
- Eischen, C.M.; Roussel, M.F.; Korsmeyer, S.J.; Cleveland, J.L. Bax loss impairs myc-induced apoptosis and circumvents the selection of p53 mutations during myc-mediated lymphomagenesis. Mol. Cell. Biol. 2001, 21, 7653–7662. [Google Scholar] [CrossRef]
- Muthalagu, N.; Junttila, M.R.; Wiese, K.E.; Wolf, E.; Morton, J.; Bauer, B.; Evan, G.I.; Eilers, M.; Murphy, D.J. Bim is the primary mediator of myc-induced apoptosis in multiple solid tissues. Cell Rep. 2014, 8, 1347–1353. [Google Scholar] [CrossRef]
- Kaur, M.; Cole, M.D. Myc acts via the pten tumor suppressor to elicit autoregulation and genome-wide gene repression by activation of the ezh2 methyltransferase. Cancer Res. 2013, 73, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Xu-Monette, Z.Y.; Deng, Q.; Manyam, G.C.; Tzankov, A.; Li, L.; Xia, Y.; Wang, X.X.; Zou, D.; Visco, C.; Dybkaer, K.; et al. Clinical and biologic significance of myc genetic mutations in de novo diffuse large b-cell lymphoma. Clin. Cancer Res. 2016, 22, 3593–3605. [Google Scholar] [CrossRef] [PubMed]
- Ennishi, D.; Mottok, A.; Ben-Neriah, S.; Shulha, H.P.; Farinha, P.; Chan, F.C.; Meissner, B.; Boyle, M.; Hother, C.; Kridel, R.; et al. Genetic profiling of myc and bcl2 in diffuse large b-cell lymphoma determines cell-of-origin-specific clinical impact. Blood 2017, 129, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, Y.; Olopade, O.I. Myc and breast cancer. Genes Cancer 2010, 1, 629–640. [Google Scholar] [CrossRef]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer alterations of the myc oncogene and its proximal network across the cancer genome atlas. Cell Syst. 2018, 6, 282–300. [Google Scholar] [CrossRef] [PubMed]
- Yuneva, M.O.; Fan, T.W.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Mates, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with myc. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Gallant, P. Myc/max/mad in invertebrates: The evolution of the max network. Curr. Top. Microbiol. Immunol. 2006, 302, 235–253. [Google Scholar]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Erisman, M.D.; Rothberg, P.G.; Diehl, R.E.; Morse, C.C.; Spandorfer, J.M.; Astrin, S.M. Deregulation of c-myc gene expression in human colon carcinoma is not accompanied by amplification or rearrangement of the gene. Mol. Cell. Biol. 1985, 5, 1969. [Google Scholar] [CrossRef] [PubMed]
- Sikora, K.; Chan, S.; Evan, G.; Gabra, H.; Markham, N.; Stewart, J.; Watson, J. C-myc oncogene expression in colorectal cancer. Cancer 1987, 59, 1289–1295. [Google Scholar] [CrossRef]
- Van de Wetering, M.; Sancho, E.; Verweij, C.; de Lau, W.; Oving, I.; Hurlstone, A.; van der Horn, K.; Batlle, E.; Coudreuse, D.; Haramis, A.P.; et al. The beta-catenin/tcf-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [Google Scholar] [CrossRef]
- Sansom, O.J.; Meniel, V.S.; Muncan, V.; Phesse, T.J.; Wilkins, J.A.; Reed, K.R.; Vass, J.K.; Athineos, D.; Clevers, H.; Clarke, A.R. Myc deletion rescues apc deficiency in the small intestine. Nature 2007, 446, 676–679. [Google Scholar] [CrossRef]
- De Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for lgr5(+) stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef]
- Bu, Y.; Cao, D. The origin of cancer stem cells. Front. Biosci. (Sch. Ed.) 2012, 4, 819–830. [Google Scholar]
- Shen, Y.; Cao, D. Hepatocellular carcinoma stem cells: Origins and roles in hepatocarcinogenesis and disease progression. Front. Biosci. (Elite Ed.) 2012, 4, 1157–1169. [Google Scholar] [CrossRef]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of cd133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
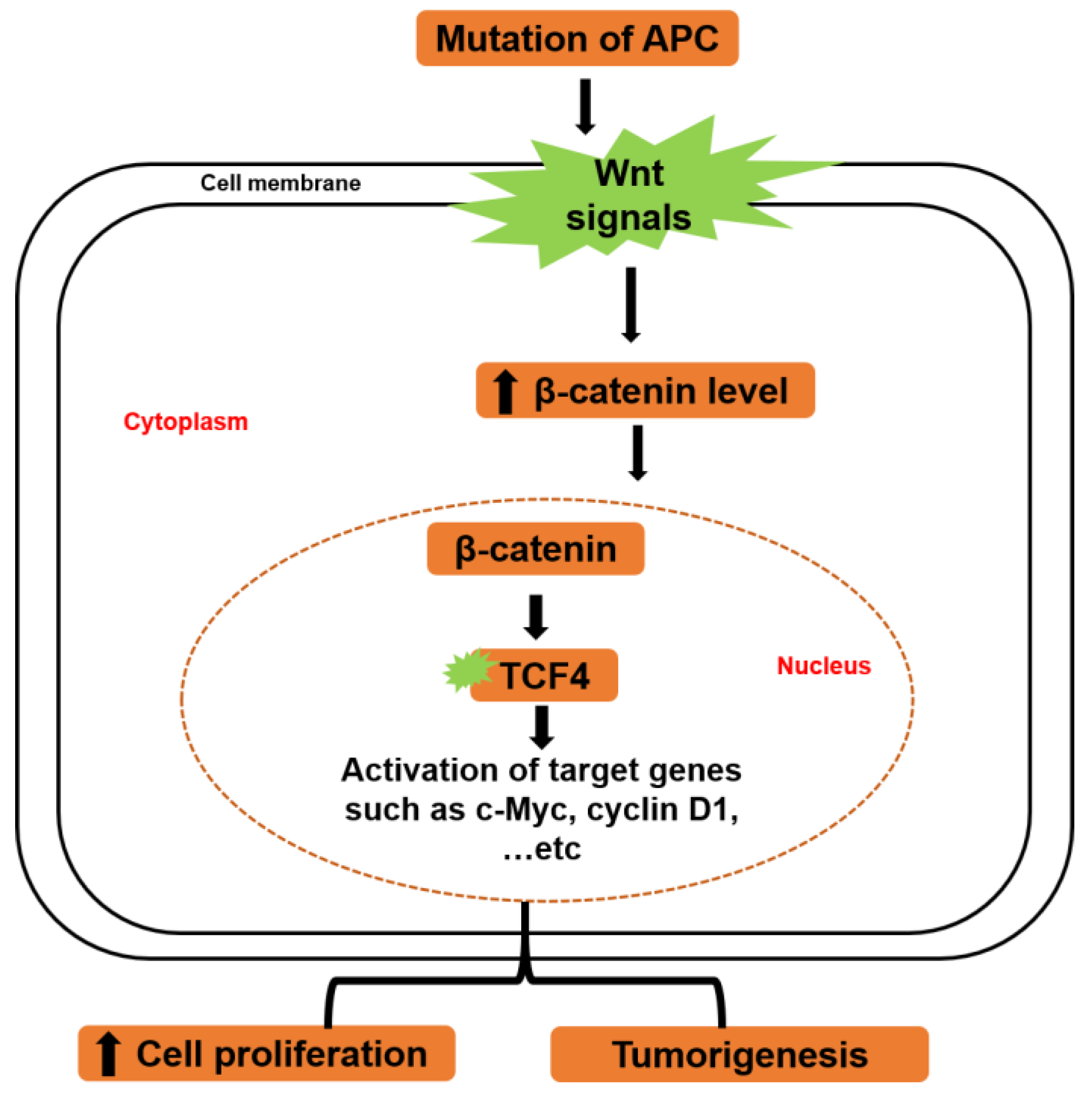
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-myc as a target of the apc pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Powell, S.M.; Zilz, N.; Beazer-Barclay, Y.; Bryan, T.M.; Hamilton, S.R.; Thibodeau, S.N.; Vogelstein, B.; Kinzler, K.W. Apc mutations occur early during colorectal tumorigenesis. Nature 1992, 359, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.K.; Griffith, O.L.; Tai, I.T.; Jones, S.J. Meta-analysis of colorectal cancer gene expression profiling studies identifies consistently reported candidate biomarkers. Cancer Epidemiol. Biomark. Prev. 2008, 17, 543–552. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-tcf signaling in colon cancer by mutations in beta-catenin or apc. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef]
- Morin, P.J. Beta-catenin signaling and cancer. Bioessays 1999, 21, 1021–1030. [Google Scholar] [CrossRef]
- De Sousa, E.M.F.; Colak, S.; Buikhuisen, J.; Koster, J.; Cameron, K.; de Jong, J.H.; Tuynman, J.B.; Prasetyanti, P.R.; Fessler, E.; van den Bergh, S.P.; et al. Methylation of cancer-stem-cell-associated wnt target genes predicts poor prognosis in colorectal cancer patients. Cell Stem Cell 2011, 9, 476–485. [Google Scholar]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [Green Version]
- De Sousa, E.M.F.; Wang, X.; Jansen, M.; Fessler, E.; Trinh, A.; de Rooij, L.P.; de Jong, J.H.; de Boer, O.J.; van Leersum, R.; Bijlsma, M.F.; et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat. Med. 2013, 19, 614–618. [Google Scholar] [CrossRef]
- Toon, C.W.; Chou, A.; Clarkson, A.; DeSilva, K.; Houang, M.; Chan, J.C.; Sioson, L.L.; Jankova, L.; Gill, A.J. Immunohistochemistry for myc predicts survival in colorectal cancer. Plos ONE 2014, 9, e87456. [Google Scholar] [CrossRef]
- Chen, Z.; He, X.; Jia, M.; Liu, Y.; Qu, D.; Wu, D.; Wu, P.; Ni, C.; Zhang, Z.; Ye, J.; et al. Beta-catenin overexpression in the nucleus predicts progress disease and unfavourable survival in colorectal cancer: A meta-analysis. Plos ONE 2013, 8, e63854. [Google Scholar]
- Qiao, L.; Wong, B.C.Y. Role of notch signaling in colorectal cancer. Carcinogenesis 2009, 30, 1979–1986. [Google Scholar] [CrossRef]
- Parsons, D.W.; Wang, T.L.; Samuels, Y.; Bardelli, A.; Cummins, J.M.; DeLong, L.; Silliman, N.; Ptak, J.; Szabo, S.; Willson, J.K.; et al. Colorectal cancer: Mutations in a signalling pathway. Nature 2005, 436, 792. [Google Scholar] [CrossRef]
- Alves-Guerra, M.C.; Ronchini, C.; Capobianco, A.J. Mastermind-like 1 is a specific coactivator of beta-catenin transcription activation and is essential for colon carcinoma cell survival. Cancer Res. 2007, 67, 8690–8698. [Google Scholar] [CrossRef]
- Meng, R.D.; Shelton, C.C.; Li, Y.M.; Qin, L.X.; Notterman, D.; Paty, P.B.; Schwartz, G.K. Gamma-secretase inhibitors abrogate oxaliplatin-induced activation of the notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res. 2009, 69, 573–582. [Google Scholar] [CrossRef]
- Yao, J.; Duan, L.; Fan, M.; Yuan, J.; Wu, X. Notch1 induces cell cycle arrest and apoptosis in human cervical cancer cells: Involvement of nuclear factor kappa b inhibition. Int. J. Gynecol. Cancer 2007, 17, 502–510. [Google Scholar] [CrossRef]
- Qi, R.; An, H.; Yu, Y.; Zhang, M.; Liu, S.; Xu, H.; Guo, Z.; Cheng, T.; Cao, X. Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res. 2003, 63, 8323–8329. [Google Scholar]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz i Altaba, A. Human colon cancer epithelial cells harbour active hedgehog-gli signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. Embo Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef]
- Akiyoshi, T.; Nakamura, M.; Koga, K.; Nakashima, H.; Yao, T.; Tsuneyoshi, M.; Tanaka, M.; Katano, M. Gli1, downregulated in colorectal cancers, inhibits proliferation of colon cancer cells involving wnt signalling activation. Gut 2006, 55, 991–999. [Google Scholar] [CrossRef]
- Kerbel, R.S.; Kobayashi, H.; Graham, C.H. Intrinsic or acquired drug resistance and metastasis: Are they linked phenotypes? J. Cell. Biochem. 1994, 56, 37–47. [Google Scholar] [CrossRef]
- Dijt, F.J.; Fichtinger-Schepman, A.M.; Berends, F.; Reedijk, J. Formation and repair of cisplatin-induced adducts to DNA in cultured normal and repair-deficient human fibroblasts. Cancer Res. 1988, 48, 6058–6062. [Google Scholar] [PubMed]
- Dabholkar, M.; Bostick-Bruton, F.; Weber, C.; Bohr, V.A.; Egwuagu, C.; Reed, E. Ercc1 and ercc2 expression in malignant tissues from ovarian cancer patients. J. Natl. Cancer Inst. 1992, 84, 1512–1517. [Google Scholar] [CrossRef]
- Sheridan, E.; Silcocks, P.; Smith, J.; Hancock, B.W.; Goyns, M.H. P53 mutation in a series of epithelial ovarian cancers from the u.K., and its prognostic significance. Eur. J. Cancer 1994, 30a, 1701–1704. [Google Scholar] [CrossRef]
- Perego, P.; Giarola, M.; Righetti, S.C.; Supino, R.; Caserini, C.; Delia, D.; Pierotti, M.A.; Miyashita, T.; Reed, J.C.; Zunino, F. Association between cisplatin resistance and mutation of p53 gene and reduced bax expression in ovarian carcinoma cell systems. Cancer Res. 1996, 56, 556–562. [Google Scholar]
- Agarwal, R.; Kaye, S.B. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A family of drug transporters: The multidrug resistance-associated proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
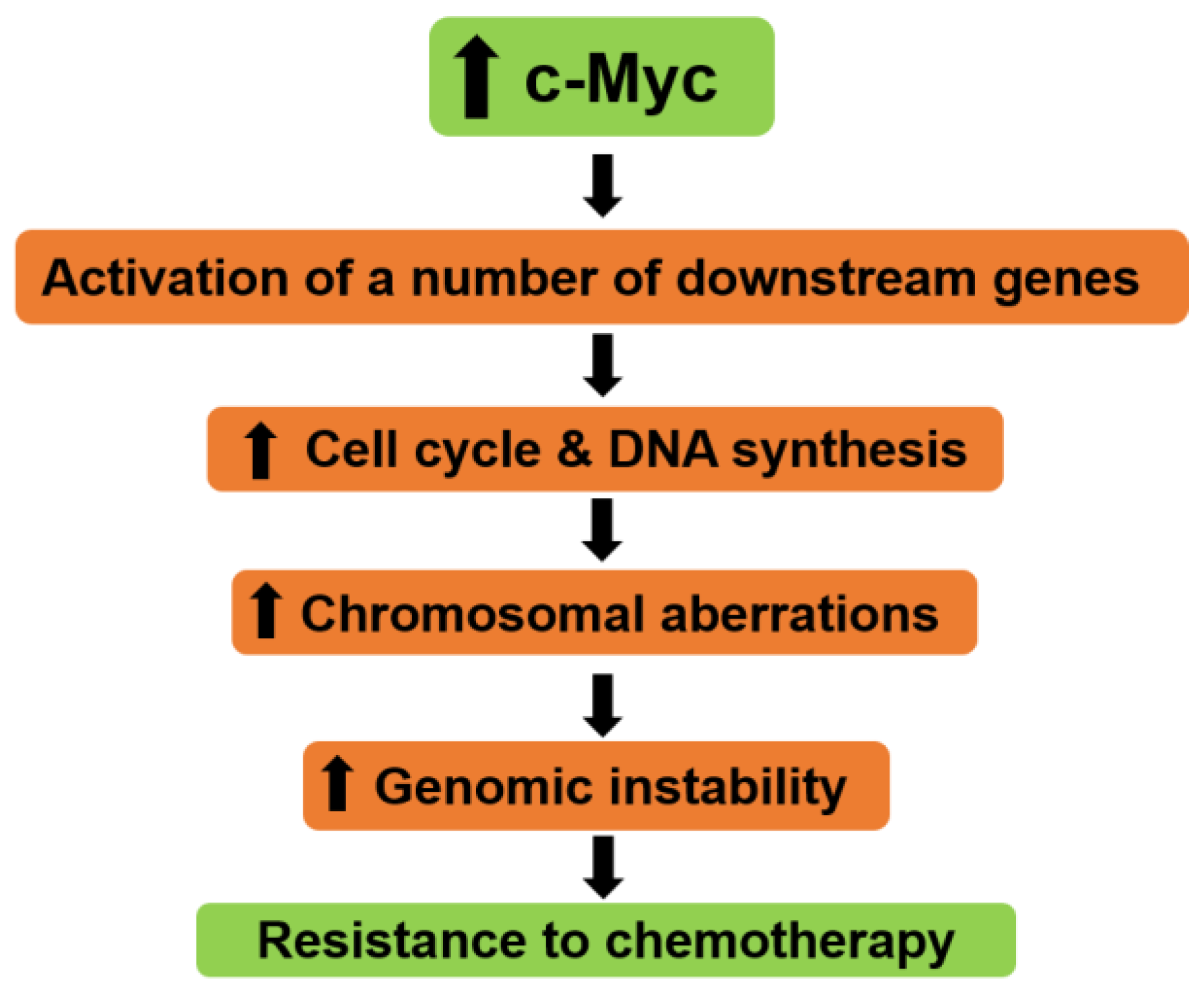
- Neiman, P.E.; Kimmel, R.; Icreverzi, A.; Elsaesser, K.; Bowers, S.J.; Burnside, J.; Delrow, J. Genomic instability during myc-induced lymphomagenesis in the bursa of fabricius. Oncogene 2006, 25, 6325–6335. [Google Scholar] [CrossRef] [PubMed]
- Prochownik, E.V. C-myc: Linking transformation and genomic instability. Curr. Mol. Med. 2008, 8, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Kuzyk, A.; Mai, S. C-myc-induced genomic instability. Cold Spring Harb. Perspect. Med. 2014, 4, a014373. [Google Scholar] [CrossRef]
- Kuttler, F.; Mai, S. Formation of non-random extrachromosomal elements during development, differentiation and oncogenesis. Semin. Cancer Biol. 2007, 17, 56–64. [Google Scholar] [CrossRef]
- Mai, S.; Fluri, M.; Siwarski, D.; Huppi, K. Genomic instability in mycer-activated rat1a-mycer cells. Chromosome Res. 1996, 4, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Felsher, D.W.; Bishop, J.M. Transient excess of myc activity can elicit genomic instability and tumorigenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 3940–3944. [Google Scholar] [CrossRef]
- Rockwood, L.D.; Torrey, T.A.; Kim, J.S.; Coleman, A.E.; Kovalchuk, A.L.; Xiang, S.; Ried, T.; Morse, H.C., 3rd; Janz, S. Genomic instability in mouse burkitt lymphoma is dominated by illegitimate genetic recombinations, not point mutations. Oncogene 2002, 21, 7235–7240. [Google Scholar] [CrossRef]
- Silva, A.G.; Graves, H.A.; Guffei, A.; Ricca, T.I.; Mortara, R.A.; Jasiulionis, M.G.; Mai, S. Telomere-centromere-driven genomic instability contributes to karyotype evolution in a mouse model of melanoma. Neoplasia 2010, 12, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gomez-Gonzalez, B. Genome instability: A mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008, 9, 204–217. [Google Scholar] [CrossRef]
- Rebucci, M.; Michiels, C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem. Pharm. 2013, 85, 1219–1226. [Google Scholar] [CrossRef]
- Pyndiah, S.; Tanida, S.; Ahmed, K.M.; Cassimere, E.K.; Choe, C.; Sakamuro, D. C-myc suppresses bin1 to release poly(adp-ribose) polymerase 1: A mechanism by which cancer cells acquire cisplatin resistance. Sci. Signal. 2011, 4, ra19. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.L.; White, J.D.; Esdale, W.J.; Burton, M.A.; DeCruz, E.E. Tumour cells surviving in vivo cisplatin chemotherapy display elevated c-myc expression. Br. J. Cancer 1996, 73, 610–614. [Google Scholar] [CrossRef] [Green Version]
- Knapp, D.C.; Mata, J.E.; Reddy, M.T.; Devi, G.R.; Iversen, P.L. Resistance to chemotherapeutic drugs overcome by c-myc inhibition in a lewis lung carcinoma murine model. Anticancer Drugs 2003, 14, 39–47. [Google Scholar] [CrossRef]
- Lin, C.P.; Liu, J.D.; Chow, J.M.; Liu, C.R.; Liu, H.E. Small-molecule c-myc inhibitor, 10058-f4, inhibits proliferation, downregulates human telomerase reverse transcriptase and enhances chemosensitivity in human hepatocellular carcinoma cells. Anticancer Drugs 2007, 18, 161–170. [Google Scholar] [CrossRef]
- Reyes-Gonzalez, J.M.; Armaiz-Pena, G.N.; Mangala, L.S.; Valiyeva, F.; Ivan, C.; Pradeep, S.; Echevarria-Vargas, I.M.; Rivera-Reyes, A.; Sood, A.K.; Vivas-Mejia, P.E. Targeting c-myc in platinum-resistant ovarian cancer. Mol. Cancer 2015, 14, 2260–2269. [Google Scholar] [CrossRef]
- Gravina, G.L.; Festuccia, C.; Popov, V.M.; Di Rocco, A.; Colapietro, A.; Sanita, P.; Monache, S.D.; Musio, D.; De Felice, F.; Di Cesare, E.; et al. C-myc sustains transformed phenotype and promotes radioresistance of embryonal rhabdomyosarcoma cell lines. Radiat. Res. 2016, 185, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, C.; Biroccio, A.; Candiloro, A.; Citro, G.; Fornari, C.; Mottolese, M.; Del Bufalo, D.; Zupi, G. Increase of cisplatin sensitivity by c-myc antisense oligodeoxynucleotides in a human metastatic melanoma inherently resistant to cisplatin. Clin. Cancer Res. 1999, 5, 2588–2595. [Google Scholar] [PubMed]
- Huang, M.J.; Cheng, Y.C.; Liu, C.R.; Lin, S.; Liu, H.E. A small-molecule c-myc inhibitor, 10058-f4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp. Hematol. 2006, 34, 1480–1489. [Google Scholar] [CrossRef]
- Zhang, H.L.; Wang, P.; Lu, M.Z.; Zhang, S.D.; Zheng, L. c-Myc maintains the self-renewal and chemoresistance properties of colon cancer stem cells. Oncol. Lett. 2019, 17, 4487–4493. [Google Scholar] [CrossRef]
- Bartfeld, S.; Clevers, H. Stem cell-derived organoids and their application for medical research and patient treatment. J. Mol. Med. 2017, 95, 729–738. [Google Scholar] [CrossRef]
- Kim, M.J.; Xia, B.; Suh, H.N.; Lee, S.H.; Jun, S.; Lien, E.M.; Zhang, J.; Chen, K.; Park, J.I. Paf-myc-controlled cell stemness is required for intestinal regeneration and tumorigenesis. Dev. Cell 2018, 44, 582–596.e584. [Google Scholar] [CrossRef]
- Usui, T.; Sakurai, M.; Umata, K.; Yamawaki, H.; Ohama, T.; Sato, K. Preparation of human primary colon tissue-derived organoid using air liquid interface culture. Curr. Protoc. Toxicol. 2018, 75, 22.6.1–22.6.7. [Google Scholar]
- Usui, T.; Sakurai, M.; Enjoji, S.; Kawasaki, H.; Umata, K.; Ohama, T.; Fujiwara, N.; Yabe, R.; Tsuji, S.; Yamawaki, H.; et al. Establishment of a novel model for anticancer drug resistance in three-dimensional primary culture of tumor microenvironment. Stem. Cells Int. 2016, 2016, 7053872. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Batsaikhan, B.E.; Yoshikawa, K.; Kurita, N.; Iwata, T.; Takasu, C.; Kashihara, H.; Shimada, M. Cyclopamine decreased the expression of sonic hedgehog and its downstream genes in colon cancer stem cells. Anticancer Res. 2014, 34, 6339–6344. [Google Scholar]
- Usui, T.; Sakurai, M.; Umata, K.; Elbadawy, M.; Ohama, T.; Yamawaki, H.; Hazama, S.; Takenouchi, H.; Nakajima, M.; Tsunedomi, R.; et al. Hedgehog signals mediate anti-cancer drug resistance in three-dimensional primary colorectal cancer organoid culture. Int. J. Mol. Sci. 2018, 19, 1098. [Google Scholar] [CrossRef] [PubMed]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Development of an experimental model for analyzing drug resistance in colorectal cancer. Cancers 2018, 10, 164. [Google Scholar] [CrossRef]
- Duong, H.Q.; Nemazanyy, I.; Rambow, F.; Tang, S.C.; Delaunay, S.; Tharun, L.; Florin, A.; Buttner, R.; Vandaele, D.; Close, P.; et al. The endosomal protein cemip links wnt signaling to mek1-erk1/2 activation in selumetinib-resistant intestinal organoids. Cancer Res. 2018, 78, 4533–4548. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Banister, C.E.; Weige, C.C.; Altomare, D.; Richardson, J.H.; Contreras, C.M.; Buckhaults, P.J. Prdm1 silences stem cell-related genes and inhibits proliferation of human colon tumor organoids. Proc. Natl. Acad. Sci. USA 2018, 115, E5066–E5075. [Google Scholar] [CrossRef]
- Kang, H.B.; Lee, H.R.; Jee da, J.; Shin, S.H.; Nah, S.S.; Yoon, S.Y.; Kim, J.W. Prdm1, a tumor-suppressor gene, is induced by genkwadaphnin in human colon cancer sw620 cells. J. Cell Biochem. 2016, 117, 172–179. [Google Scholar] [CrossRef]
- Felsher, D.W.; Bishop, J.M. Reversible tumorigenesis by myc in hematopoietic lineages. Mol. Cell 1999, 4, 199–207. [Google Scholar] [CrossRef]
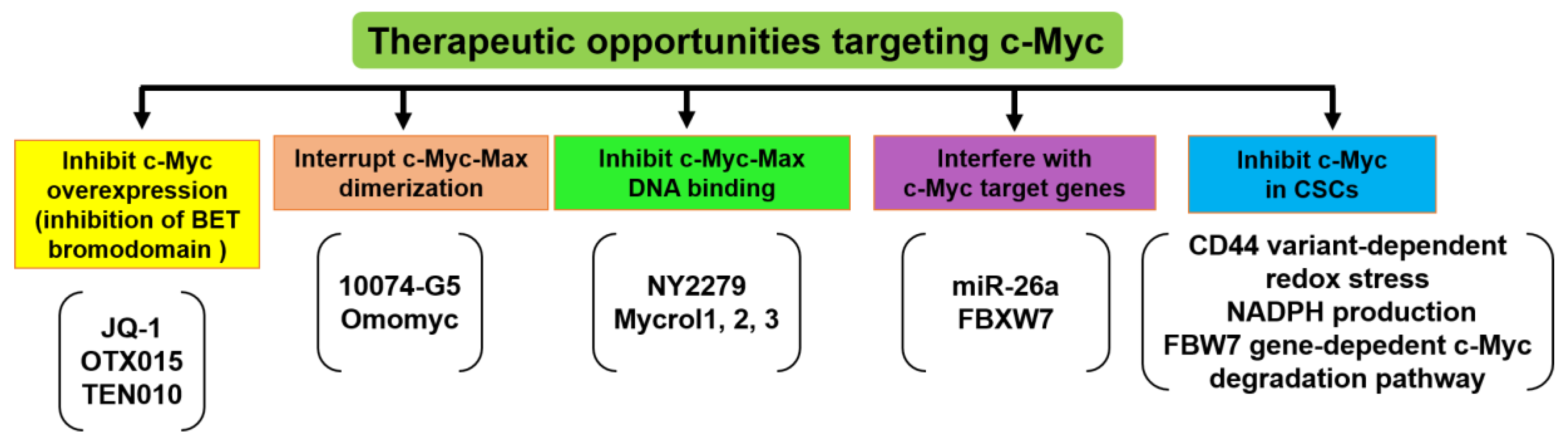
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., 3rd. Targeting myc dependence in cancer by inhibiting bet bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [PubMed]
- Clausen, D.M.; Guo, J.; Parise, R.A.; Beumer, J.H.; Egorin, M.J.; Lazo, J.S.; Prochownik, E.V.; Eiseman, J.L. In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-g5, a novel small-molecule inhibitor of c-myc/max dimerization. J. Pharm. Exp. 2010, 335, 715–727. [Google Scholar] [CrossRef]
- Follis, A.V.; Hammoudeh, D.I.; Daab, A.T.; Metallo, S.J. Small-molecule perturbation of competing interactions between c-myc and max. Bioorg. Med. Chem. Lett. 2009, 19, 807–810. [Google Scholar] [CrossRef]
- Hammoudeh, D.I.; Follis, A.V.; Prochownik, E.V.; Metallo, S.J. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-myc. J. Am. Chem. Soc. 2009, 131, 7390–7401. [Google Scholar] [CrossRef]
- Prochownik, E.V.; Vogt, P.K. Therapeutic targeting of myc. Genes Cancer 2010, 1, 650–659. [Google Scholar] [CrossRef]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Chang, T.C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microrna delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef]
- Frenzel, A.; Loven, J.; Henriksson, M.A. Targeting myc-regulated mirnas to combat cancer. Genes Cancer 2010, 1, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Loven, J.; Zinin, N.; Wahlstrom, T.; Muller, I.; Brodin, P.; Fredlund, E.; Ribacke, U.; Pivarcsi, A.; Pahlman, S.; Henriksson, M. Mycn-regulated micrornas repress estrogen receptor-alpha (esr1) expression and neuronal differentiation in human neuroblastoma. Proc. Natl. Acad. Sci. USA 2010, 107, 1553–1558. [Google Scholar] [CrossRef]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of ldh-a expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase a induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef]
- Farrell, A.S.; Sears, R.C. Myc degradation. Cold Spring Harb. Perspect. Med. 2014, 4, a014365. [Google Scholar] [CrossRef]
- Rajagopalan, H.; Jallepalli, P.V.; Rago, C.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Inactivation of hcdc4 can cause chromosomal instability. Nature 2004, 428, 77–81. [Google Scholar] [CrossRef]
- Diefenbacher, M.E.; Popov, N.; Blake, S.M.; Schulein-Volk, C.; Nye, E.; Spencer-Dene, B.; Jaenicke, L.A.; Eilers, M.; Behrens, A. The deubiquitinase usp28 controls intestinal homeostasis and promotes colorectal cancer. J. Clin. Invest. 2014, 124, 3407–3418. [Google Scholar] [CrossRef]
- Yoshida, G.J. Emerging roles of myc in stem cell biology and novel tumor therapies. J. Exp. Clin. Cancer Res. 2018, 37, 173. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Emerging Roles of C-Myc in Cancer Stem Cell-Related Signaling and Resistance to Cancer Chemotherapy: A Potential Therapeutic Target Against Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 2340. https://doi.org/10.3390/ijms20092340
Elbadawy M, Usui T, Yamawaki H, Sasaki K. Emerging Roles of C-Myc in Cancer Stem Cell-Related Signaling and Resistance to Cancer Chemotherapy: A Potential Therapeutic Target Against Colorectal Cancer. International Journal of Molecular Sciences. 2019; 20(9):2340. https://doi.org/10.3390/ijms20092340
Chicago/Turabian StyleElbadawy, Mohamed, Tatsuya Usui, Hideyuki Yamawaki, and Kazuaki Sasaki. 2019. "Emerging Roles of C-Myc in Cancer Stem Cell-Related Signaling and Resistance to Cancer Chemotherapy: A Potential Therapeutic Target Against Colorectal Cancer" International Journal of Molecular Sciences 20, no. 9: 2340. https://doi.org/10.3390/ijms20092340