Cisplatin Synergistically Enhances Antitumor Potency of Conditionally Replicating Adenovirus via p53 Dependent or Independent Pathways in Human Lung Carcinoma


Abstract
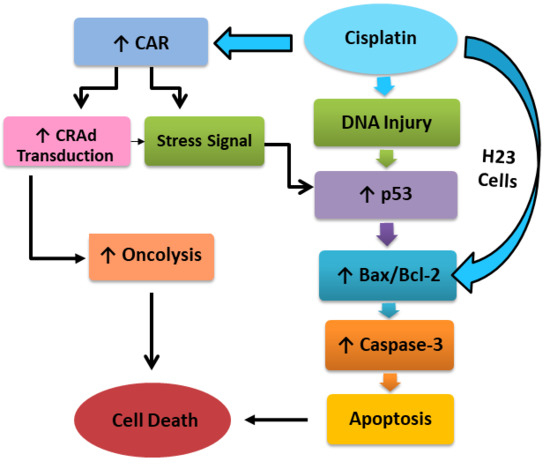
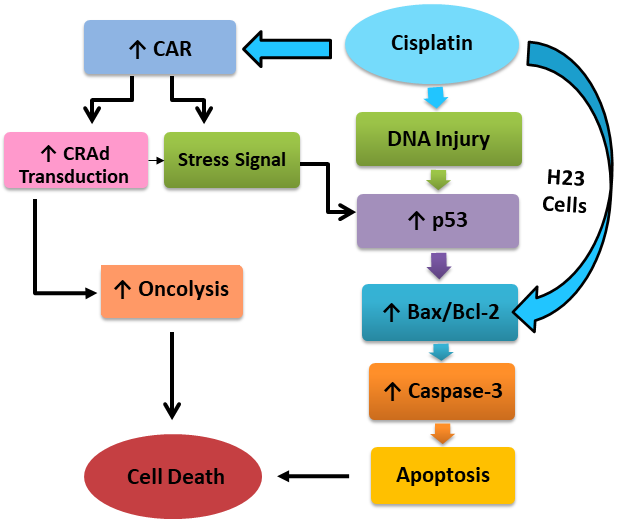
:
1. Introduction
2. Results
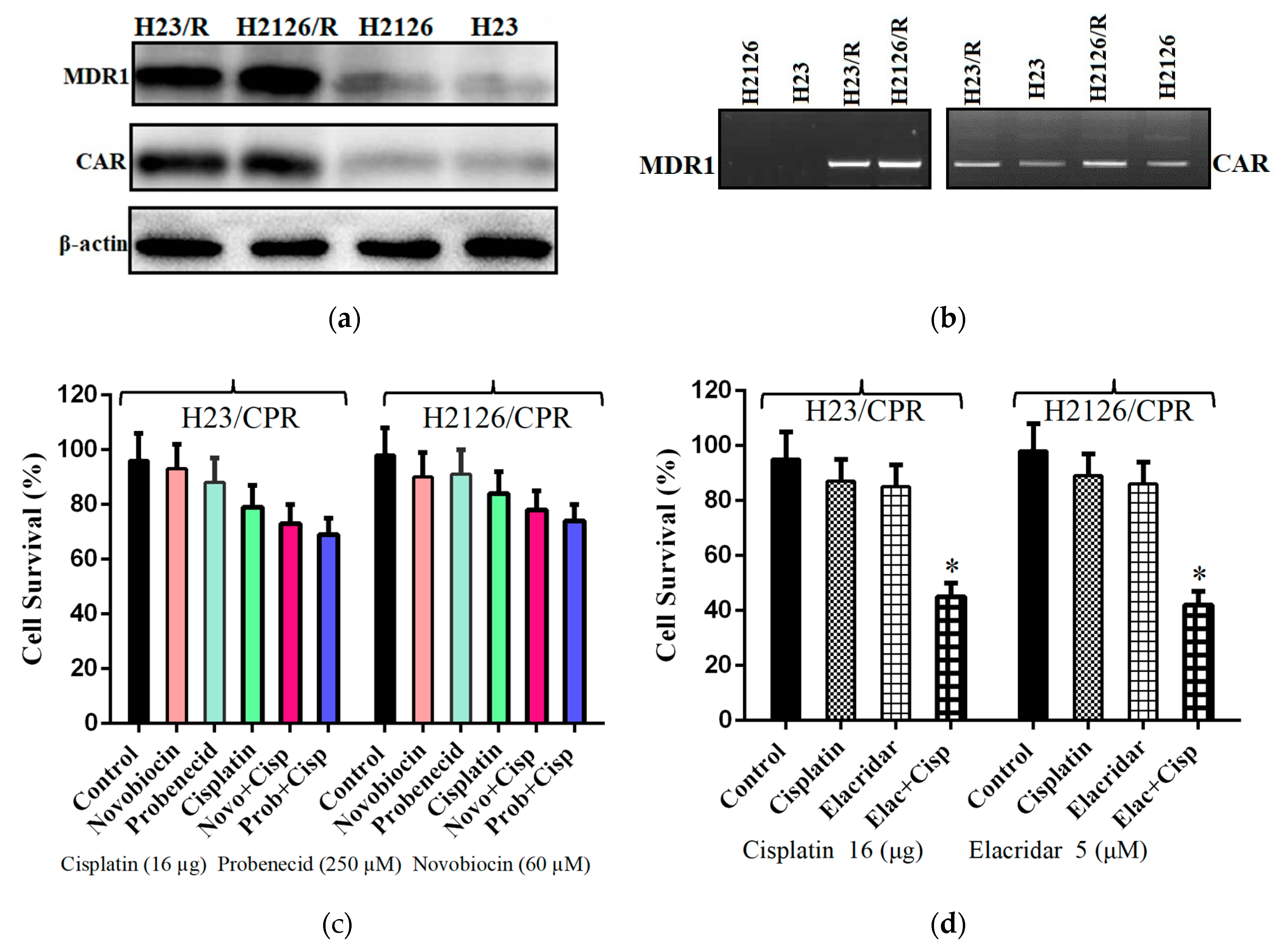
2.1. Multidrug Resistance and CAR in Cisplatin Resistant Lung Cancer Cells
2.2. Cisplatin-Resistant Lung Cancer Cells are More Sensitive to Adenoviral Infection with CRAd
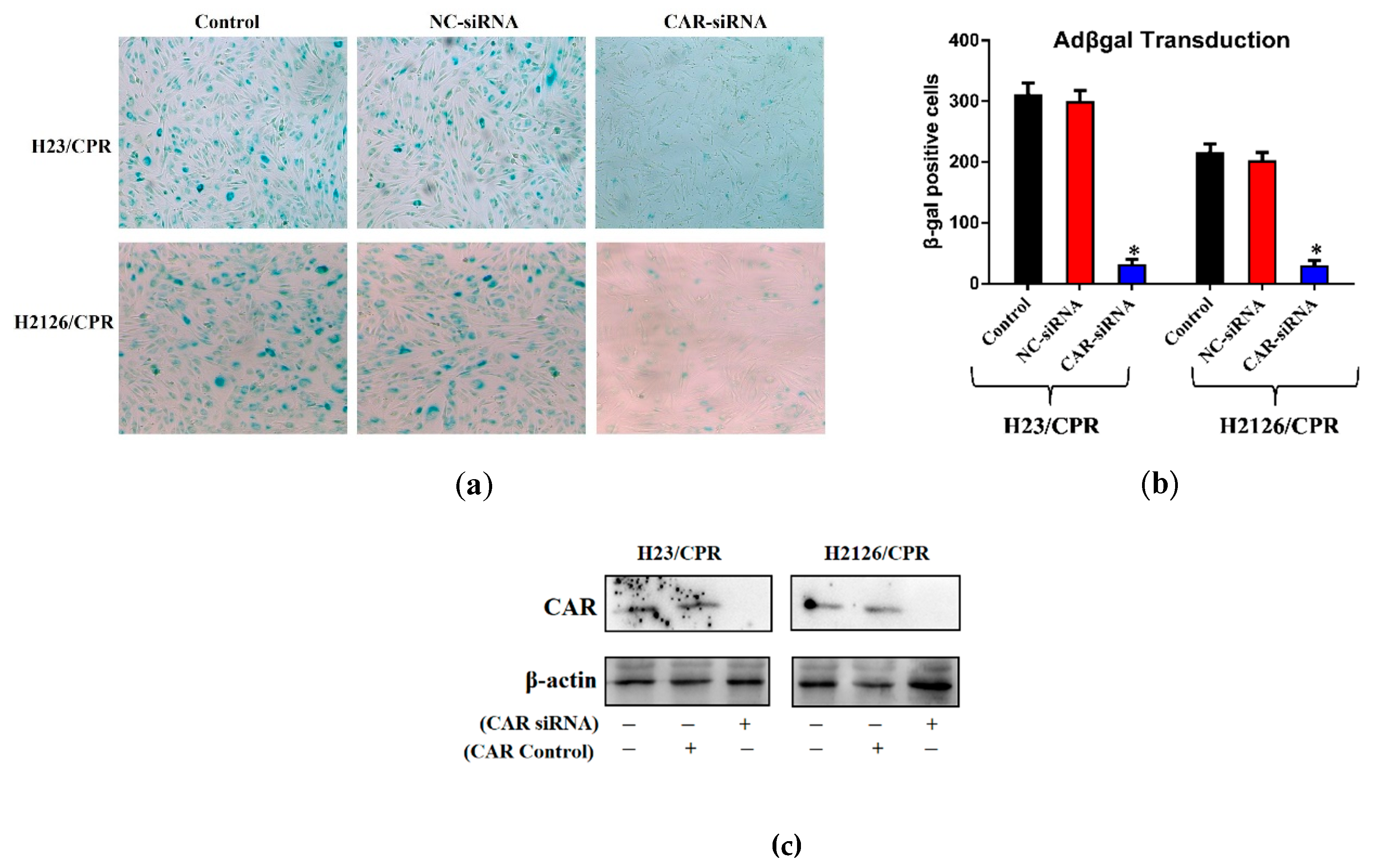
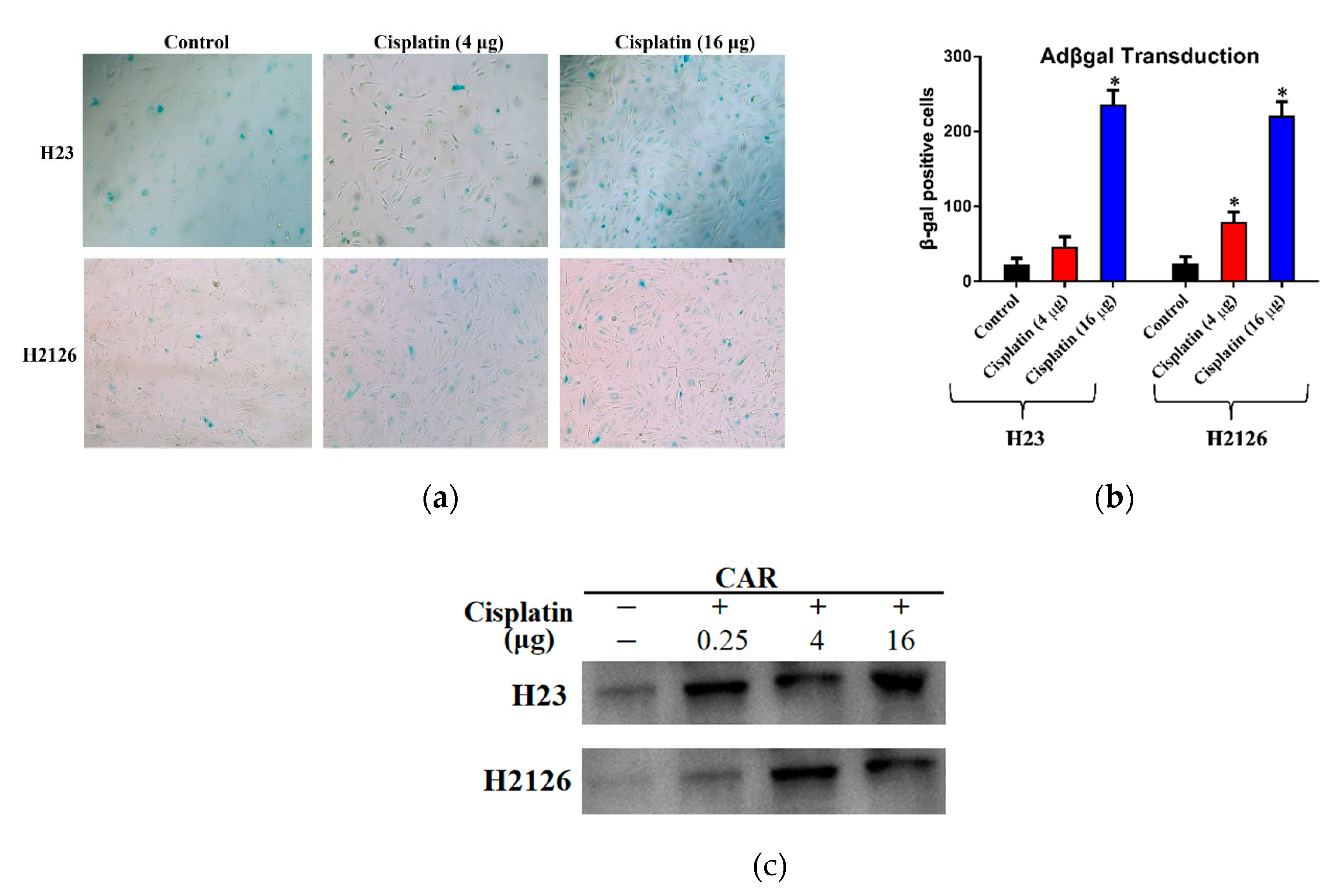
2.3. Cisplatin-Mediated CAR Upregulation is Responsible for Raised Transduction Insensitive Cells
2.4. Synergistic Antitumor Effect of Cisplatin and CRAd in Cancer Cells
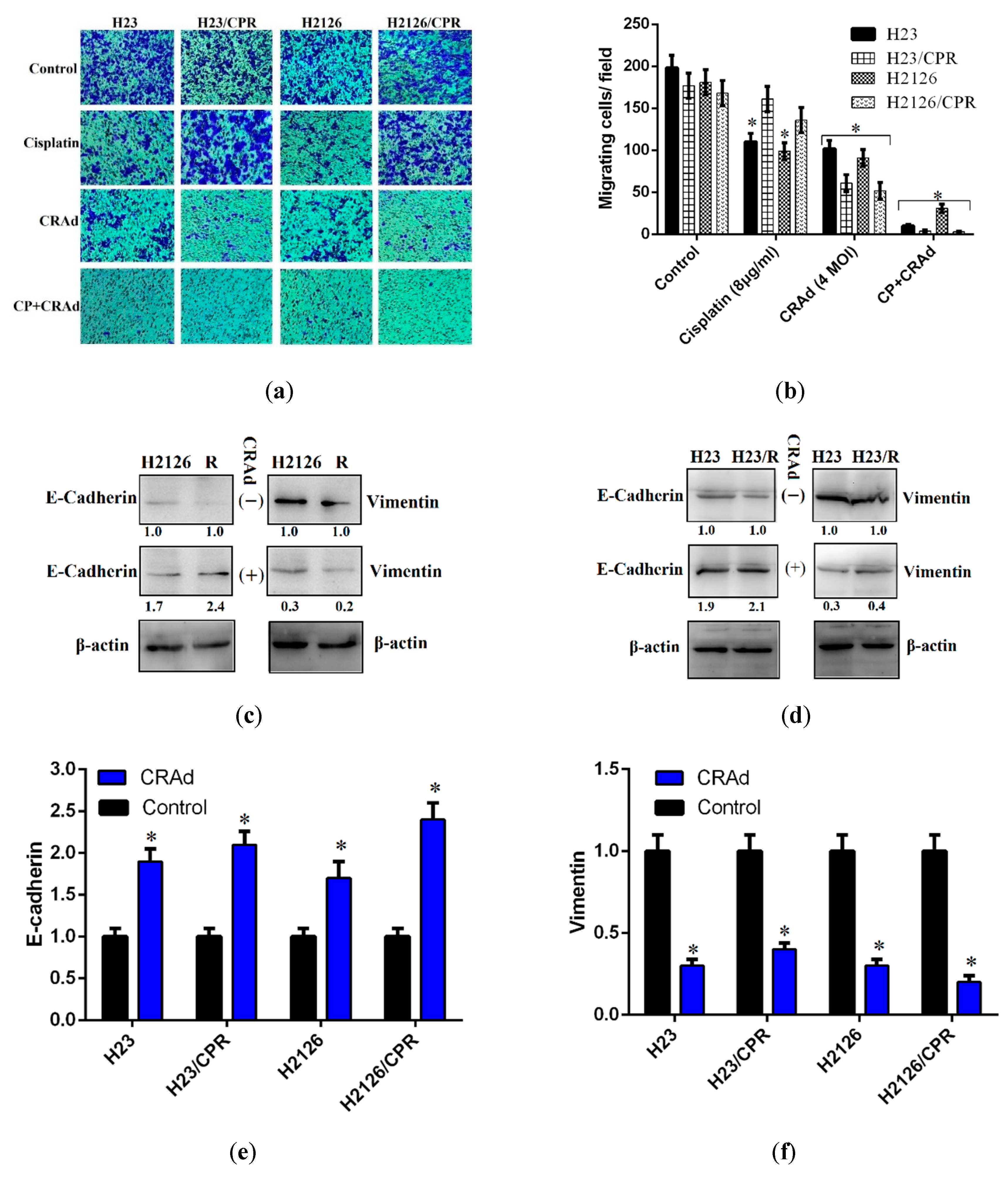
2.5. Cisplatin and CRAd Inhibited Cancer Cell Migration
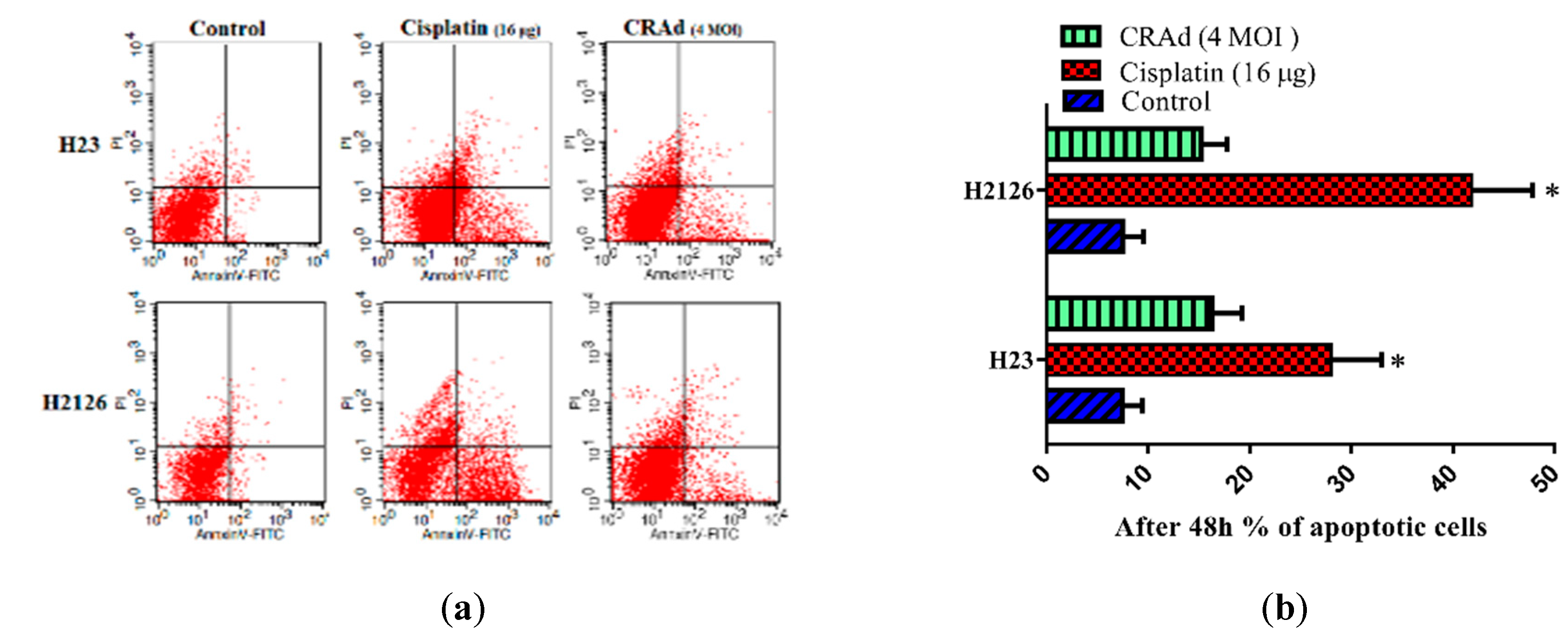
2.6. Cisplatin and CRAd Induce Apoptosis in Lung Cancer Cells by Activating the Caspase Pathway
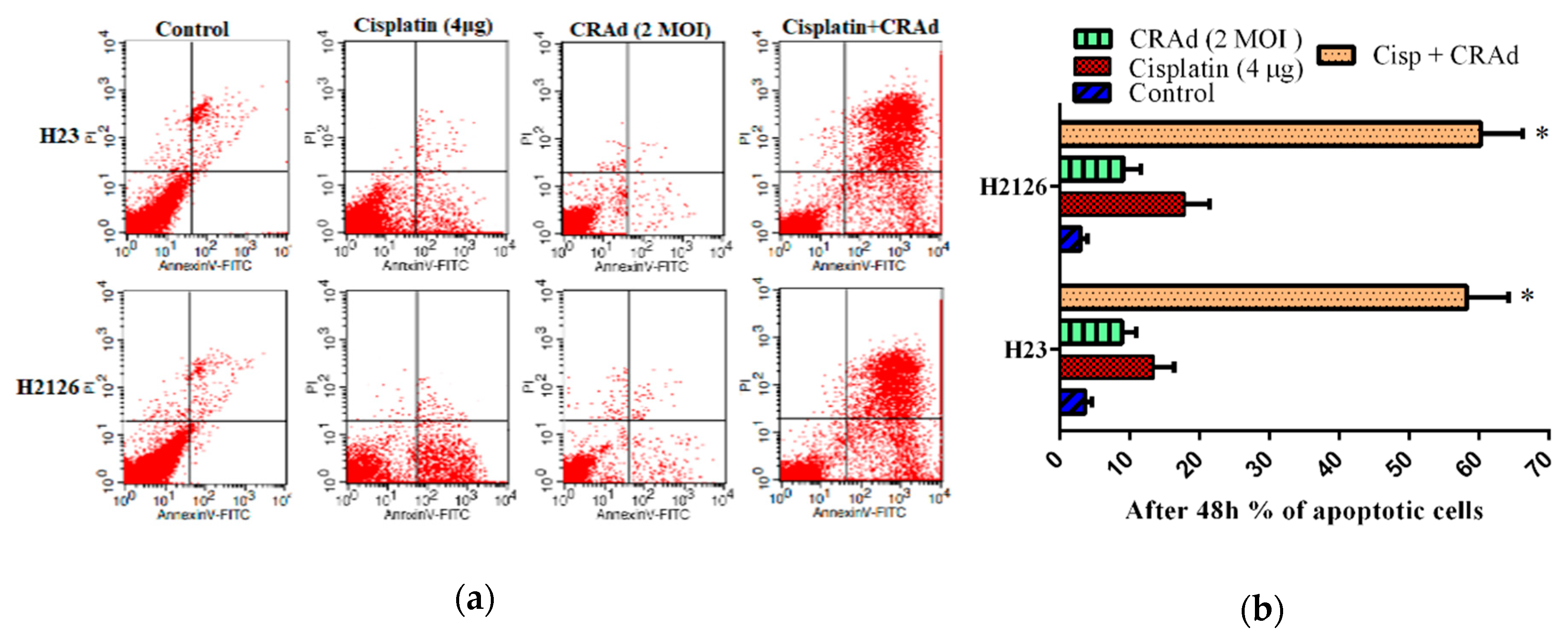
2.7. Co-Treatment of Cisplatin with CRAd Promotes Apoptosis Dependent and Independent Death of Lung Adenocarcinoma Cells in a Synergistic Manner
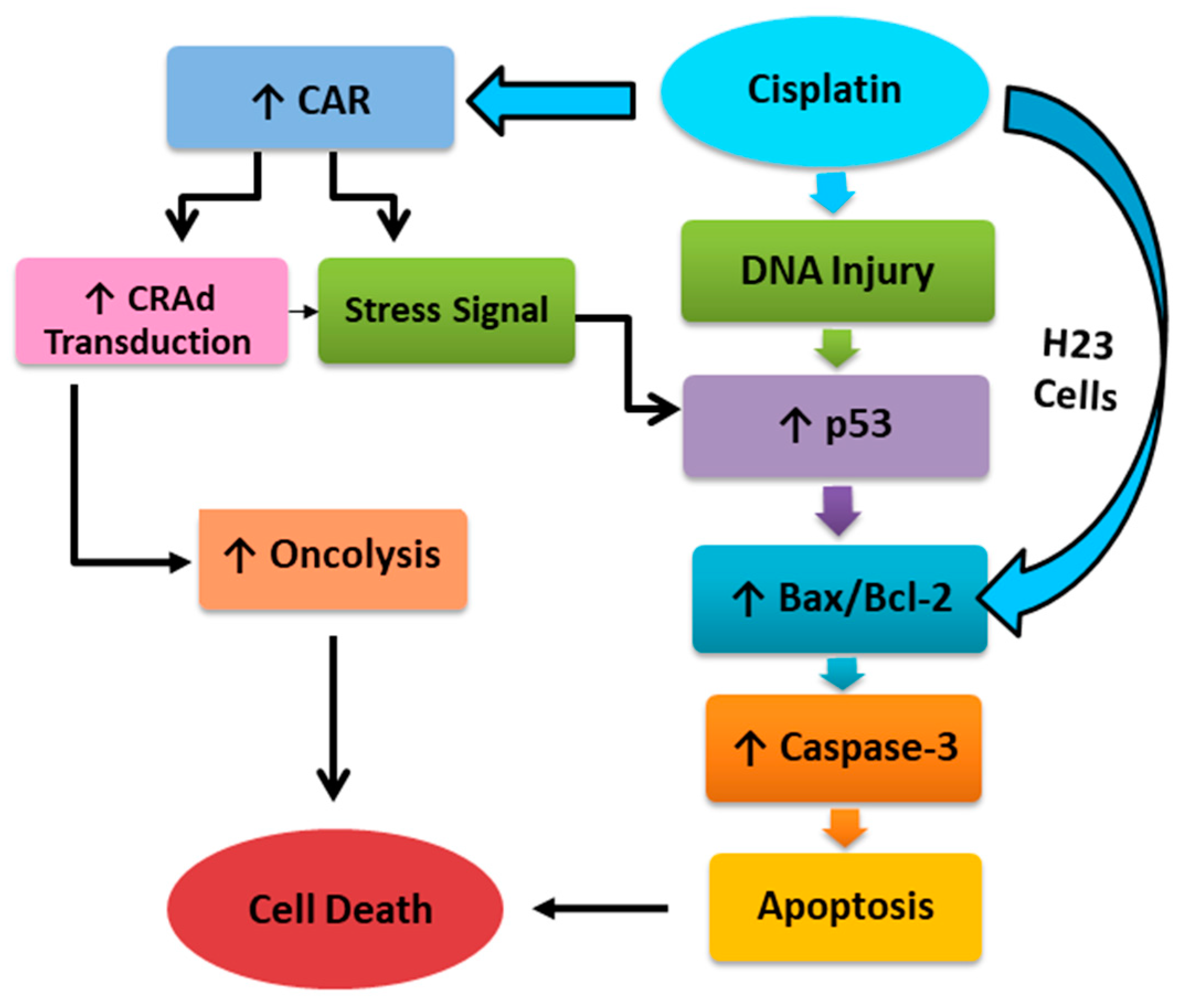
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Adv Transduction Assessment via β-Gal Activity
4.3. siRNA and Transfection
4.4. Western Blotting
4.5. Reverse Transcription-qPCR
4.6. MTT Assay
4.7. In Vitro Cell Migration Assay
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAR | Coxsackievirus and adenovirus receptor |
| CRAd | Conditionally Replicating Adenovirus |
| Bcl-2 | B-cell lymphoma 2 |
| Bax | Bcl-2-associated X |
| EMT | Epithelial to mesenchymal transition |
| CPR | Cisplatin resistant |
| RT-PCR | Reverse transcription polymerase chain reaction |
| MTT | (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) tetrazolium |
| MDR1 | Multidrug resistance gene 1 |
| MDR | Multidrug resistance |
| PCR | Polymerase chain reaction |
| DMSO | Dimethyl sulfoxide |
| DDPR | Diamminedichloroplatinum resistant |
| DMEM | Dulbecco’s modified eagle’s medium |
| FBS | Fetal bovine serum |
| EMT | Epithelial–mesenchymal transition |
| RIPA | Radioimmunoprecipitation assay buffer |
| PVDF | Polyvinylidene difluoride |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Novello, S.; Barlesi, F.; Califano, R.; Cufer, T.; Ekman, S.; Levra, M.G.; Kerr, K.; Popat, S.; Reck, M.; Senan, S.; et al. Metastatic non-small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27 (Suppl. 5), v1–v27. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Tabata, M.; Kiura, K.; Kozuki, T.; Hisamoto, A.; Katayama, H.; Takigawa, N.; Fujimoto, N.; Fujiwara, K.; Ueoka, H.; et al. Gefitinib induces premature senescence in non-small cell lung cancer cells with or without EGFR gene mutation. Oncol. Rep. 2007, 17, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012, 30, 679. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Jänne, P.A. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012, 2, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Pastan, I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993, 62, 385–427. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.J.; dos Santos, D.J.; Ferreira, M.J.U. P-glycoprotein and membrane roles in multidrug resistance. Future Med. Chem. 2015, 7, 929–946. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Chen, H.L.; Carbone, D.P. Gene therapy for lung cancer. Ann. Oncol. 1995, 6 (Suppl. 3), S61–S63. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, K.; Kobayashi, D.; Furuya, D.; Tsuji, N.; Yagihashi, A.; Watanabe, N. A role for survivin in radioresistance of pancreatic cancer cells. Jpn. J. Cancer Res. 2002, 93, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Kaneko, K.; Hirota, M.; Masamune, A.; Satoh, A.; Shimosegawa, T. Expression of survivin is correlated with cancer cell apoptosis and is involved in the development of human pancreatic duct cell tumors. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2001, 92, 271–278. [Google Scholar] [CrossRef]
- Bao, R.; Connolly, D.C.; Murphy, M.; Green, J.; Weinstein, J.K.; Pisarcik, D.A.; Hamilton, T.C. Activation of cancer-specific gene expression by the survivin promoter. J. Natl. Cancer Inst. 2002, 94, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Fengzhi, L.I.; Altieri, D.C. Transcriptional analysis of human survivin gene expression. Biochem. J. 1999, 344, 305–311. [Google Scholar]
- Raschperger, E.; Thyberg, J.; Pettersson, S.; Philipson, L.; Fuxe, J.; Pettersson, R.F. The coxsackie-and adenovirus receptor (CAR) is an in vivo marker for epithelial tight junctions, with a potential role in regulating permeability and tissue homeostasis. Exp. Cell Res. 2006, 312, 1566–1580. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhao, J.; Lu, J.; Jiang, Y.; Yang, H.; Li, P.; Zhao, M.; Liu, K.; Dong, Z. Coxsackievirus and adenovirus receptor promotes antitumor activity of oncolytic adenovirus H101 in esophageal cancer. Int. J. Mol. Med. 2012, 30, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Kasala, D.; Na, Y.; Lee, M.S.; Kim, S.W.; Jeong, J.H.; Yun, C.O. Enhanced therapeutic efficacy of an adenovirus-PEI-bile-acid complex in tumors with low coxsackie and adenovirus receptor expression. Biomaterials 2014, 35, 5505–5516. [Google Scholar] [CrossRef] [PubMed]
- Ingemarsdotter, C.K.; Tookman, L.A.; Browne, A.; Pirlo, K.; Cutts, R.; Chelela, C.; Khurrum, K.F.; Leung, E.Y.; Dowson, S.; Webber, L.; et al. Paclitaxel resistance increases oncolytic adenovirus efficacy via upregulated CAR expression and dysfunctional cell cycle control. Mol. Oncol. 2015, 9, 791–805. [Google Scholar] [CrossRef] [PubMed]
- Yanan, L.; Sakhawat, A.; Ma, L.; Wang, S.; Huang, Y. Improved Antitumor Effect of Survivin Responsive Conditional Replication Adenovirus in Combination with Cisplatin in Lung Cancer. J. Cancer Sci. Ther. 2016, 8, 216–221. [Google Scholar] [CrossRef]
- Yang, C.-H.; Chen, Y.-C.; Kuo, M.-L. Novobiocin sensitizes BCRP/MXR/ABCP overexpressing topotecan-resistant human breast carcinoma cells to topotecan and mitoxantrone. Anticancer Res. 2003, 23, 2519–2523. [Google Scholar] [PubMed]
- Issandou, M.; Grand-Perret, T. Multidrug resistance P-glycoprotein is not involved in cholesterol esterifcation. Biochem. Biophys. Res. Commun. 2000, 279, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Ambriović-Ristov, A.; Gabrilovac, J.; Čimbora-Zovko, T.; Osmak, M. Increased adenoviral transduction efficacy in human laryngeal carcinoma cells resistant to cisplatin is associated with increased expression of integrin αvβ3 and coxsackie adenovirus receptor. Int. J. Cancer 2004, 110, 660–667. [Google Scholar] [PubMed]
- Sakhawat, A.; Liu, Y.; Ling Ma, T.M.; Wang, S.; Zhang, L.; Cong, X.; Huang, Y. Upregulation of Coxsackie Adenovirus Receptor Sensitizes Cisplatin-Resistant Lung Cancer Cells to CRAd-Induced Inhibition. J. Cancer 2017, 8, 1425. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Yano, S.; Tazawa, H.; Yoshida, R.; Urata, Y.; Kagawa, S.; Fujiwara, T. Biological Effects of Oncolytic Adenovirus on Epithelial-Mesenchymal Transition in Human Lung Cancer Cells. Mol. Ther. 2011, 19, S306. [Google Scholar] [CrossRef]
- Rosenberg, B.; Vancamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385. [Google Scholar] [CrossRef] [PubMed]
- Pignon, J.P.; Tribodet, H.; Scagliotti, G.V.; Douillard, J.Y.; Shepherd, F.A.; Stephens, R.J.; Dunant, A.; Torri, V.; Rosell, R.; Seymour, L.; et al. Lung adjuvant cisplatin evaluation: A pooled analysis by the LACE Collaborative Group. J. Clin. Oncol. 2008, 26, 3552–3559. [Google Scholar] [CrossRef] [PubMed]
- Vale, C. Advanced Bladder Cancer (ABC) Meta-analysis Collaboration. Neoadjuvant chemotherapy in invasive bladder cancer: A systematic review and meta-analysis. Lancet 2003, 361, 1927–1934. [Google Scholar] [CrossRef]
- Sandler, A.B.; Nemunaitis, J.; Denham, C.; Von Pawel, J.; Cormier, Y.; Gatzemeier, U.; Mattson, K.; Manegold, C.; Palmer, M.C.; Gregor, A.; et al. Phase III trial of gemcitabine plus cisplatin versus cisplatin alone in patients with locally advanced or metastatic non–small-cell lung cancer. J. Clin. Oncol. 2000, 18, 122. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.H.; Harrington, D.; Belani, C.P.; Langer, C.; Sandler, A.; Krook, J.; Zhu, J.; Johnson, D.H. Comparison of four chemotherapy regimens for advanced non–small-cell lung cancer. N. Engl. J. Med. 2002, 346, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Stecker, K.; Koschel, A.; Wiedenmann, B.; Anders, M. Loss of Coxsackie and adenovirus receptor downregulates α-catenin expression. Br. J. Cancer 2009, 101, 1574. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.H.; Horng, C.T.; Lee, C.F.; Chiang, N.N.; Tsai, F.J.; Lu, C.C.; Chiang, J.H.; Hsu, Y.M.; Yang, J.S.; Chen, F.A. Epigallocatechin gallate sensitizes cisplatin-resistant oral cancer CAR cell apoptosis and autophagy through stimulating AKT/STAT3 pathway and suppressing multidrug resistance 1 signaling. Environ. Toxicol. 2017, 32, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial–mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Dean, D. Apoptosis, cell signaling, and human diseases: Molecular mechanisms. JAMA 2007, 298, 2200–2206. [Google Scholar] [CrossRef]
- Anders, M.; Vieth, M.; Röcken, C.; Ebert, M.; Pross, M.; Gretschel, S.; Schlag, P.M.; Wiedenmann, B.; Kemmner, W.; Höcker, M. Loss of the coxsackie and adenovirus receptor contributes to gastric cancer progression. Br. J. Cancer 2009, 100, 352. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Qi, T.; Wang, S.; Hao, M.; Sakhawat, A.; Liang, T.; Zhang, L.; Cong, X.; Huang, Y. Tet methylcytosine dioxygenase 1 promotes hypoxic gene induction and cell migration in colon cancer. J. Cell. Physiol. 2019, 234, 6286–6297. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, Q.; Sun, J.; Gu, A.; Jin, M.; Shen, Z.; Qiu, Z.; Wang, J.; Wang, X.; Zhan, Z.; et al. Expression of the coxsackie and adenovirus receptor in human lung cancers. Tumor Biol. 2013, 34, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Huo, X.; Wang, L.; Meng, Q.; Liu, Z.; Liu, Q.; Sun, H.; Sun, P.; Peng, J.; Liu, K. Dioscin strengthens the efficiency of adriamycin in MCF-7 and MCF-7/ADR cells through autophagy induction: More than just down-regulation of MDR1. Sci. Rep. 2016, 6, 28403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, D.; Kar, R.; Gupta, B.; Halder, S.; Garg, S.; Mehndiratta, M.; Wadhwa, N.; Agarwal, R. Role of Survivin and p53 Expression in Response of Primary Culture of Ovarian Cancer Cells to Treatment with Chemotherapeutic Agents. Int. J. Gynecol. Cancer 2018, 28, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Labernadie, A.; Kato, T.; Brugués, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; González-Tarragó, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 2017, 19, 224. [Google Scholar] [CrossRef] [PubMed]
- Beggs, J.E.; Tian, S.; Jones, G.G.; Xie, J.; Iadevaia, V.; Jenei, V.; Thomas, G.; Proud, C.G. The MAP kinase-interacting kinases regulate cell migration, vimentin expression and eIF4E/CYFIP1 binding. Biochem. J. 2015, 467, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Louisa, M.; Soediro, T.M.; Suyatna, F.D. In vitro modulation of P-glycoprotein, MRP-1 and BCRP expression by mangiferin in doxorubicin-treated MCF-7 cells. Asian Pac. J. Cancer. Prev. 2014, 15, 1639–1642. [Google Scholar] [CrossRef]
- Lin, H.; Yang, G.; Yu, J.; Wang, J.; Li, Q.; Guo, S.; Cao, B. KDM5c inhibits multidrug resistance of colon cancer cell line by down-regulating ABCC1. Biomed. Pharmacother. 2018, 107, 1205–1209. [Google Scholar] [CrossRef]
- Bernd, A.; Ott, M.; Ishikawa, H.; Schroten, H.; Schwerk, C.; Fricker, G. Characterization of efflux transport proteins of the human choroid plexus papilloma cell line HIBCPP, a functional in vitro model of the blood-cerebrospinal fluid barrier. Pharm. Res. 2015, 32, 2973–2982. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.C.; Huang, A.C.; Wu, P.P.; Lin, H.Y.; Chueh, F.S.; Yang, J.S.; Lu, C.C.; Chiang, J.H.; Meng, M.; Chung, J.G. Gallic acid suppresses the migration and invasion of PC-3 human prostate cancer cells via inhibition of matrix metalloproteinase-2 and-9 signaling pathways. Oncol. Rep. 2011, 26, 177–184. [Google Scholar]
- Chu, C.N.; Wu, K.C.; Chung, W.S.; Zheng, L.C.; Juan, T.K.; Hsiao, Y.T.; Peng, S.F.; Yang, J.L.; Ma, Y.S.; Wu, R.S.C.; et al. Etomidate Suppresses Invasion and Migration of Human A549 Lung Adenocarcinoma Cells. Anticancer Res. 2019, 39, 215–223. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer-F | Primer-R | Ref. |
|---|---|---|---|
| CAR | GCAGGAGCCATTATAGGAACTTTG | GGACCCCAGGGATGAATGAT | [38] |
| GAPDH | GAAGGTGAAGGTCGGAGTC | GAAGATGGTGATGGGATTTC | [38] |
| ABCB1 or MDR1 | GGAGCCTACTTGGTGGCACATAA | TGGCATAGTCAGGAGCAAATGAAC | [39] |
| Survivin | AGAACTGGCCCTTCTTG GAGG | CTTTTTATGTTCCTCTAT GGGGTC | [40] |
| p53 | TAACAGTTCCTGCATGGGCGGC | AGGACAGGCACAAACACGCACC | [40] |
| E-Cadherin | TCACCACTGGGCTGGACCGA | TACAGCCTCCCACGCTGGGG | [41] |
| N-Cadherin | TCAAACACAGCCACGGCCGT | CGGTCTGGATGGCGAACCGT | [41] |
| Vimentin | TTCTCTGCCTCTTCCAAACTTT | CGTTGATAACCTGTCCATCTCTA | [42] |
| ABCG2 (BCRP) | TTCGGCTTGCAACAACTATG | TCCAGACACACCACGGATAA | [43] |
| ABCC1(MRP1) | CTCTATCTCTCCCGACATGACC | AGCAGACGATCCACAGCAAAA | [44] |
| ABCC2(MRP2) | CCCTGCTGTTCGATATACCAATC | TCGAGAGAATCCAGAATAGGGAC | [44] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, S.; Tahir, M.; Khan, A.A.; Chen, X.C.; Ling, M.; Huang, Y. Cisplatin Synergistically Enhances Antitumor Potency of Conditionally Replicating Adenovirus via p53 Dependent or Independent Pathways in Human Lung Carcinoma. Int. J. Mol. Sci. 2019, 20, 1125. https://doi.org/10.3390/ijms20051125
Ali S, Tahir M, Khan AA, Chen XC, Ling M, Huang Y. Cisplatin Synergistically Enhances Antitumor Potency of Conditionally Replicating Adenovirus via p53 Dependent or Independent Pathways in Human Lung Carcinoma. International Journal of Molecular Sciences. 2019; 20(5):1125. https://doi.org/10.3390/ijms20051125
Chicago/Turabian StyleAli, Sakhawat, Muhammad Tahir, Aamir Ali Khan, Xue Chai Chen, Ma Ling, and Yinghui Huang. 2019. "Cisplatin Synergistically Enhances Antitumor Potency of Conditionally Replicating Adenovirus via p53 Dependent or Independent Pathways in Human Lung Carcinoma" International Journal of Molecular Sciences 20, no. 5: 1125. https://doi.org/10.3390/ijms20051125