





Abstract
Glioblastoma (GBM) is the most common and aggressive primary brain tumor. Prognosis remains poor despite multimodal therapy. Developing alternative treatments is essential. Drugs targeting kinases within the phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) effectors of receptor tyrosine kinase (RTK) signaling represent promising candidates.
We previously developed a non-germline genetically engineered mouse model of GBM in which PI3K and MAPK are activated via Pten deletion and KrasG12D in immortalized astrocytes. Using this model, we examined the influence of drug potency on target inhibition, alternate pathway activation, efficacy, and synergism of single agent and combination therapy with inhibitors of these 2 pathways. Efficacy was then examined in GBM patient-derived xenografts (PDX) in vitro and in vivo.
PI3K and mitogen-activated protein kinase kinase (MEK) inhibitor potency was directly associated with target inhibition, alternate RTK effector activation, and efficacy in mutant murine astrocytes in vitro. The kinomes of GBM PDX and tumor samples were heterogeneous, with a subset of the latter harboring MAPK hyperactivation. Dual PI3K/MEK inhibitor treatment overcame alternate effector activation, was synergistic in vitro, and was more effective than single agent therapy in subcutaneous murine allografts. However, efficacy in orthotopic allografts was minimal. This was likely due to dose-limiting toxicity and incomplete target inhibition.
Drug potency influences PI3K/MEK inhibitor–induced target inhibition, adaptive kinome reprogramming, efficacy, and synergy. Our findings suggest that combination therapies with highly potent, brain-penetrant kinase inhibitors will be required to improve patient outcomes.
Glioblastoma is the most common malignant primary brain cancer. Effective treatments are limited. Targeted kinase inhibitors represent a promising alternative, but use of brain-penetrant drugs in combinations designed to overcome compensatory resistance mechanisms will likely be necessary to improve outcomes. Preclinical studies can aid identification of effective drug combinations. We used a non-germline genetically engineered mouse model with activating PI3K and MAPK mutations to show that drug potency influences signaling dynamics, efficacy, and synergism of PI3K and MEK inhibitors in vitro and in vivo. Our results demonstrate how drug potency influences response.
Glioblastoma (GBM) is the most common and aggressive adult primary brain tumor. Despite advances in diagnosis, specifically incorporation of both histological and molecular criteria in the 2016 classification, GBM is uniformly treated with surgery, radiation, and temozolomide chemotherapy.1 Recurrence is inevitable and leads to ~15 months of survival.2 Molecular heterogeneity of GBM has been extensively characterized using genomics. Primary GBM that arises de novo without a lower-grade antecedent has been stratified into 4 molecular subtypes characterized by mutations in 3 core signaling pathways: retinoblastoma, tumor protein 53, and receptor tyrosine kinase (RTK)/phosphoinositide 3-kinase (PI3K)/mitogen activated protein kinase (MAPK).3,4
The RTK/PI3K/MAPK pathways are mutated in 90% of GBM.3 PI3K and MAPK promote many cancer hallmarks, including survival, proliferation, and migration.5–7 PI3K is most frequently activated in GBM via mutations in its negative regulator, Pten.3,5,6 Activation of MAPK signaling is due either to activating mutations in RTK or Kras, or inactivating mutations in its negative regulator, NF1.3,8,9 RTK/PI3K/MAPK pathways are attractive therapeutic targets because of their mutation frequency and role in tumorigenesis.
Kinase inhibitors targeting the PI3K or MAPK pathways are currently approved for nonglioma tumors or are in clinical development.10,11 However, single agent kinase inhibitors have had disappointing clinical results in gliomas due to limited brain penetrance and drug resistance.12–14 Many kinase inhibitors, including the epidermal growth factor receptor inhibitors gefitinib and erlotinib, have low brain penetrance, restricting their ability to reach and suppress their biologic target.13 Additionally, intrinsic and acquired drug resistance has significantly limited the impact of kinase inhibitors in gliomas.13,15
Preclinical studies that define drug efficacy, elucidate resistance mechanisms, and confirm target modulation will aid in clinical drug development. Genetically engineered mouse (GEM) and patient-derived xenograft (PDX) models are valuable tools for these efforts because they enable direct genotype-to-phenotype comparisons and faithfully recapitulate the molecular heterogeneity of human tumors, respectively.13 We and others have shown that activating PI3K and MAPK mutations cooperate to promote gliomagenesis in preclinical models.16–20 We developed a series of non-germline (n)GEM models using cultured astrocytes immortalized via an N-terminal SV40 large T (T121, T) mutant that ablates the retinoblastoma family of pocket proteins. MAPK and PI3K were activated alone and in combination via oncogenic Kras (KrasG12D, R) and Pten deletion (P), respectively.20 We used these models to show that activated PI3K and MAPK cooperate to promote astrocyte proliferation, migration, and de-differentiation in vitro and malignant progression to rapidly fatal GBM in vivo. TRP astrocytes also displayed the phenotypic hallmarks of GBM stem cells (GSCs) and molecularly recapitulated proneural GBM.16,20 Here we utilized the TRP nGEM culture and allograft model system and GBM PDX to define the influence of drug potency on signaling dynamics, efficacy, and synergism of PI3K and MEK1/2 inhibitors (PI3Ki, MEKi).16,20
Materials and Methods
Supplementary methods, figures, and tables can be found online.
Cell Culture
TRP astrocyte cultures were established from mice with heterozygous TgGZT121 and KrasG12D and homozygous Pten mutations and maintained as previously described.20,21 The UNC Institutional Animal Care and Use Committee approved all animal studies (16–112). Established human cell lines (ECL) and TRP astrocytes were maintained as adherent cultures in serum-containing media.16,20,22–24 TRP astrocytes expressing luciferase were generated as previously described.16 PDX were maintained as non-adherent spheroids in serum-free media.25,26
Human GBM
Frozen, newly diagnosed GBM samples (N = 9) were obtained from the UNC Tissue Procurement Facility under a protocol approved by the UNC Office of Human Research Ethics (15–0923).
Cell Growth and Drug Synergism
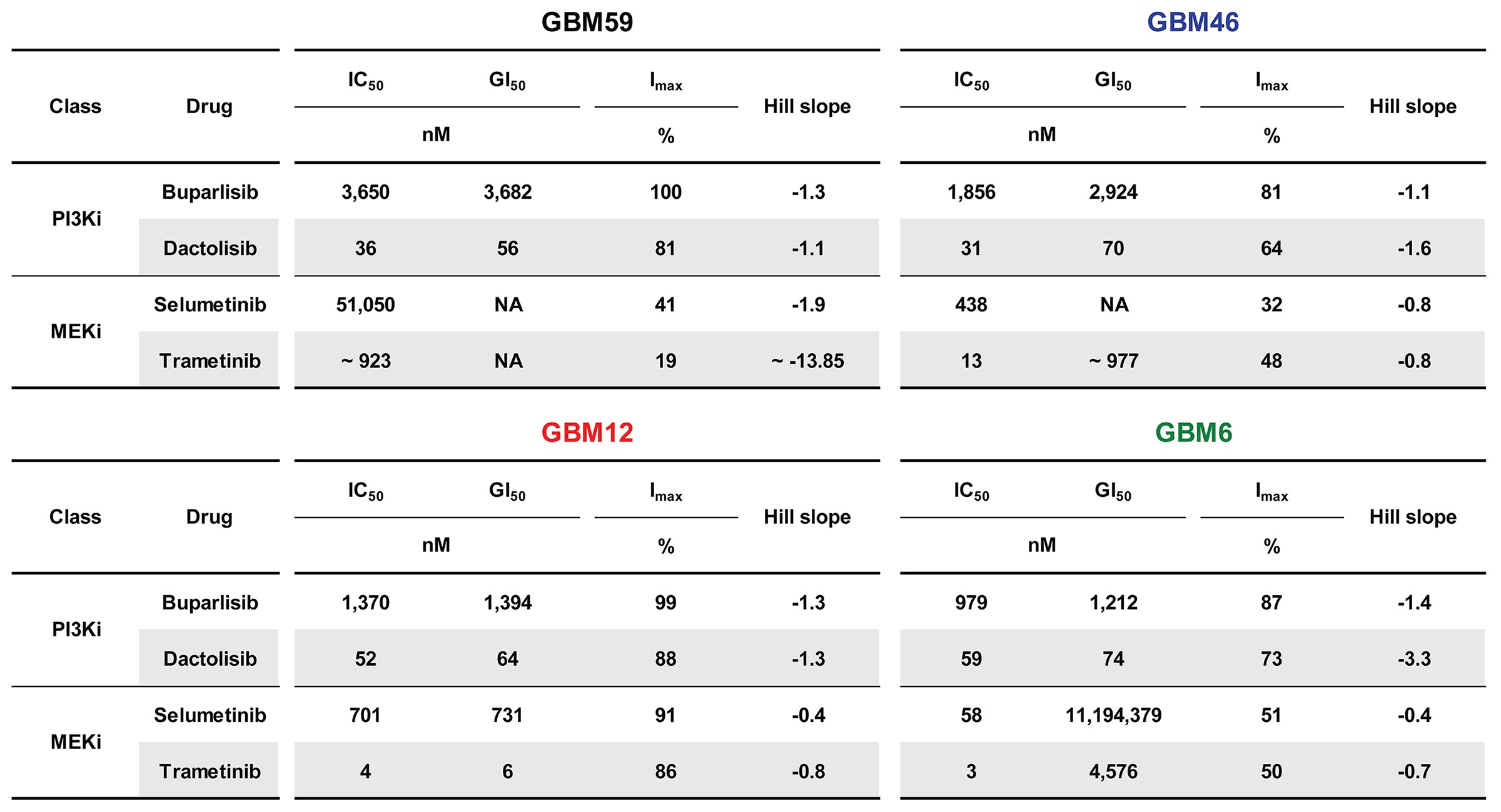
TRP cells were treated with solvent (control) or drug(s) (Supplementary Table 1) and growth was assessed with CellTiter AQ (Promega).16 PDX growth was assessed with CellTiterGlo (Promega).26 Half-maximal inhibitory concentration (IC50), 50% growth inhibition (GI50), maximum inhibition (Imax), and Hill slopes were calculated and effects of genotype and drugs on IC50 compared. PI3K/MEKi synergism was determined via the Chou–Talalay method.
Immunoblots
Proteins were extracted from cultured TRP astrocytes or allograft tumors and immunoblots were performed as previously described.16,20
Kinome Profiling
Dynamic kinome profiling was performed by multiplexed inhibitor beads and mass spectrometry (MIB-MS) on TRP astrocytes treated with buparlisib for 4–48 h. Baseline MIB-MS was performed on human PDX, ECL cultures, and GBM samples as described.27,28 Hierarchical clustering and principal components analysis were performed as described.27,28
TRP Allografts
TRP astrocytes expressing luciferase were injected orthotopically into syngeneic mice and tumor growth was monitored by bioluminescence imaging as described.16,20,24 Mice were randomized after 7 days into 4 groups and treatment was initiated on day 10 using a 5 days on/2 days off schedule until signs of neurologic morbidity (Supplementary Table S2). Mice were then sacrificed and brains harvested for immunoblots and histopathology.16,20 Alternatively, TRP astrocytes were injected into the right flank of syngeneic mice and tumors were established for 14 days. Mice were then randomized into treatment groups and treated for 5 days (Supplementary Table S2). Tumor volume was measured longitudinally for ~2 weeks. Dactolisib ± selumetinib treatments were terminated after 4 days due to drug-induced toxicity (lethargy).
Orthotopic Patient-Derived Xenografts (PDX)
PDX were established in athymic mice (Taconic) as described.29 Mice were randomized after 15 days to receive vehicle control or dactolisib. Studies were approved by the Translational Drug Development Management Animal Care and Use Committee (Scottsdale, Arizona).
Statistics and Bioinformatics
Statistics were performed in GraphPad Prism. P ≤ 0.05 was considered significant unless otherwise stated. Error bars are SEM.
Results
PI3K and MAPK mutations are frequent in GBM and drive tumorigenesis in preclinical models.3,4,8,9,30 We previously showed that activated PI3K and MAPK cooperated to promote gliomagenesis in TRP nGEM culture and allograft models.16,20 However, it remained unclear whether these models were sensitive to PI3Ki and MEKi. We addressed this issue by examining how drug potency influences target inhibition, adaptive kinome response, efficacy, and synergism of single agent and combination therapies in vitro and in vivo.
PI3Ki and MEKi Block TRP Astrocyte and PDX Growth In Vitro
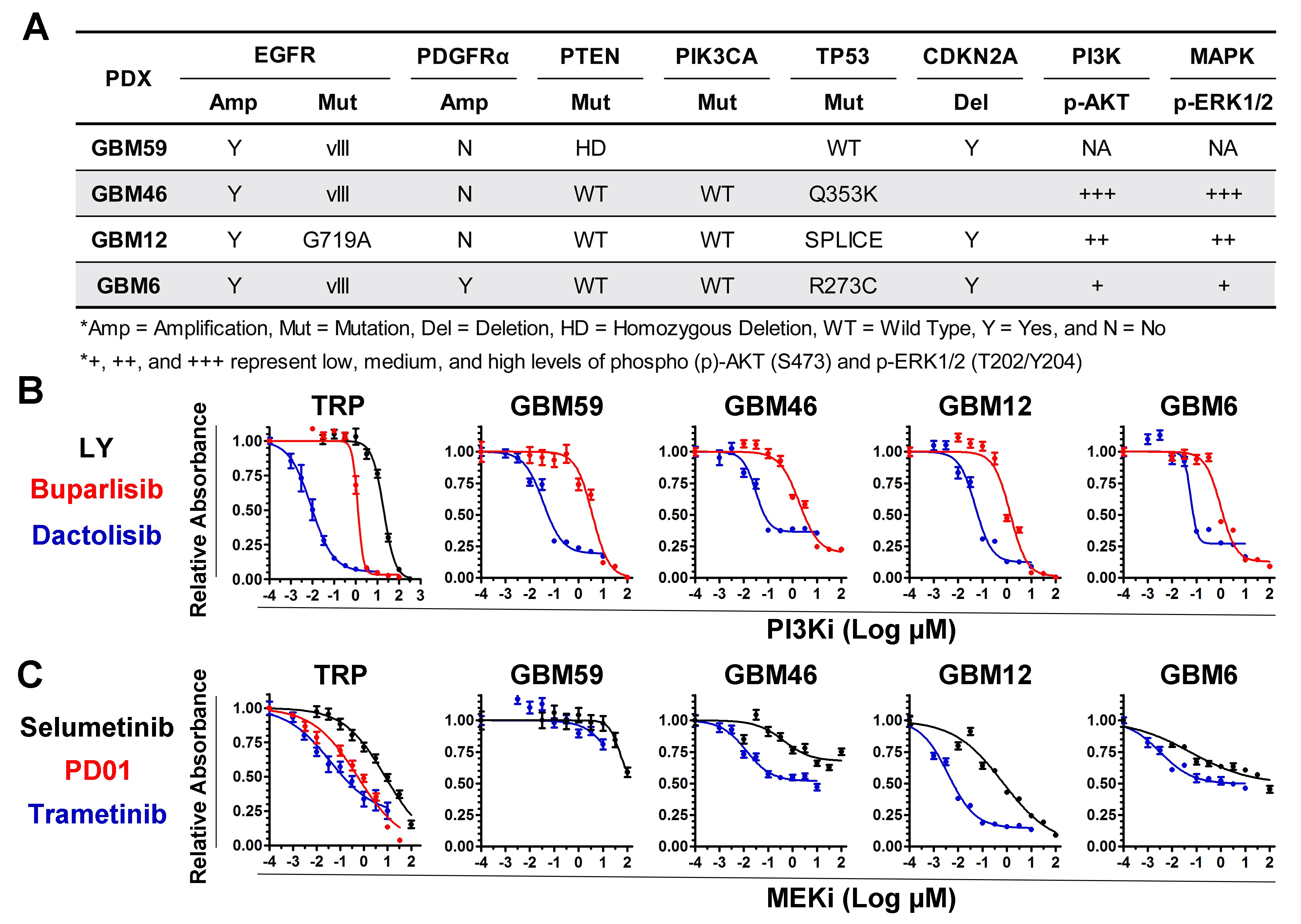
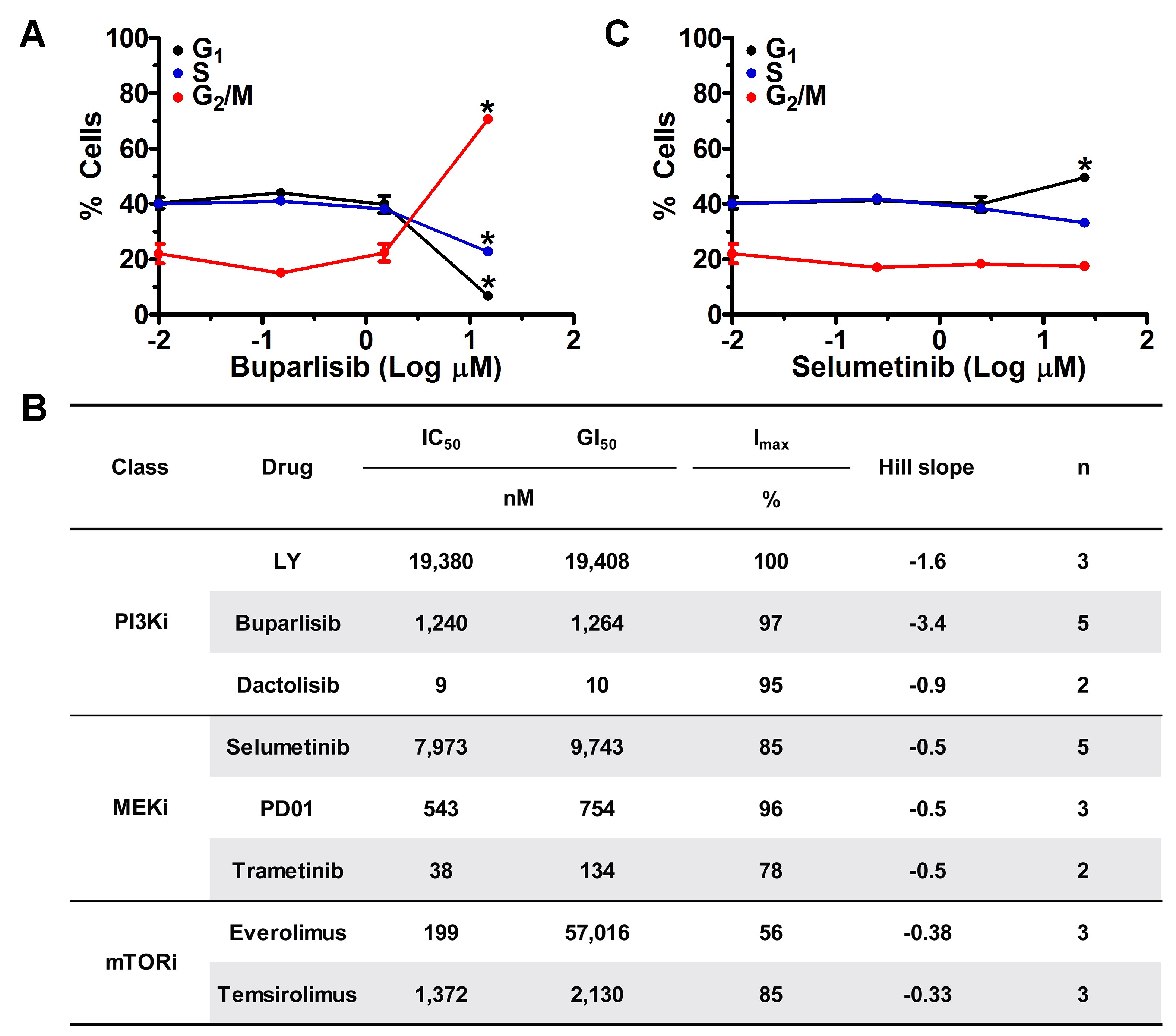
PI3Ki and MEKi efficacies were examined in cultured TRP astrocytes and epidermal growth factor receptor–amplified PDX. Both model systems have activated MAPK and PI3K signaling (Supplementary Fig. S1A).20 A dose-dependent, sigmoidal growth reduction was evident for multiple PI3Ki in these models (Supplementary Fig. S1B). The PI3Ki buparlisib induced G2/M cell cycle arrest in TRP astrocytes (Supplementary Fig. S2A). Moreover, a direct association between potency (dactolisib > buparlisib > LY294002) and efficacy was evident, both for TRP astrocytes (Fig. 1A, Supplementary Fig. S2B) and for PDX (Fig. 1B, Supplementary Fig. S3). The PI3K/mammalian target of rapamycin inhibitor (mTORi) dactolisib was the most potent, but its efficacy may be due, in part, to direct mTOR inhibition. Two mTORi reduced TRP astrocyte growth with IC50 in the low micro- to high nanomolar range (Supplementary Fig. S2B). However, both caused more gradual decreases in growth (Hill slope, P ≤ 0.03) and were less potent than dactolisib (P ≤ 0.003).
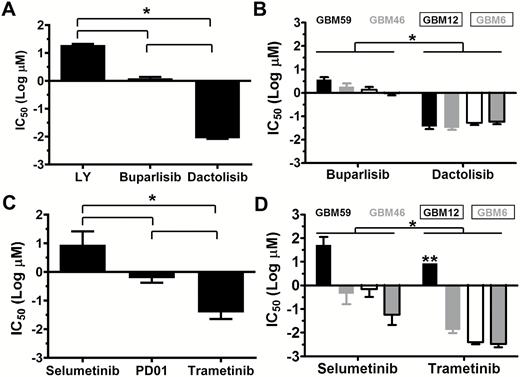
Single agent PI3Ki or MEKi potency is directly associated with efficacy in vitro. PI3Ki (AB) and MEKi (CD) potencies were inversely associated with IC50 in cultured TRP astrocytes (AC) and PDX (BD) (*P < 0.0001). **Ambiguous IC50.
Next we assessed how MEKi potency (trametinib > PD0125901 (PD01) > selumetinib) influences growth. Although PD01 and trametinib have similar IC50 for purified MEK, their mechanisms differ and trametinib more potently inhibits signaling and cell growth.31 MEKi caused gradual, dose-dependent decreases in cultured TRP and PDX growth (Supplementary Fig. S1C). Moreover, a direct association between potency and efficacy was evident, both for TRP astrocytes (Fig. 1C, Supplementary Fig. S2B) and for PDX (Fig. 1D, Supplementary Fig. S3). Selumetinib also induced G1/S cell cycle arrest in TRP astrocytes (Supplementary Fig. S2C). Taken together, these data showed that cultured TRP astrocytes and PDX were sensitive to PI3Ki and MEKi.
PI3Ki Induces Adaptive Kinome Reprogramming, Including Alternate MAPK Activation
Among PI3Ki, mTORi, and MEKi, PI3Ki were generally most effective at inhibiting TRP astrocyte growth (Fig. 1A, C, Supplementary Figures S1, S2). However, many targeted kinase inhibitors effective in preclinical settings have had disappointing results when used as single agents in GBM patients.12,13 One reason may be compensatory signaling changes that manifest as drug resistance, suggesting that combination therapies will be necessary to improve outcomes.
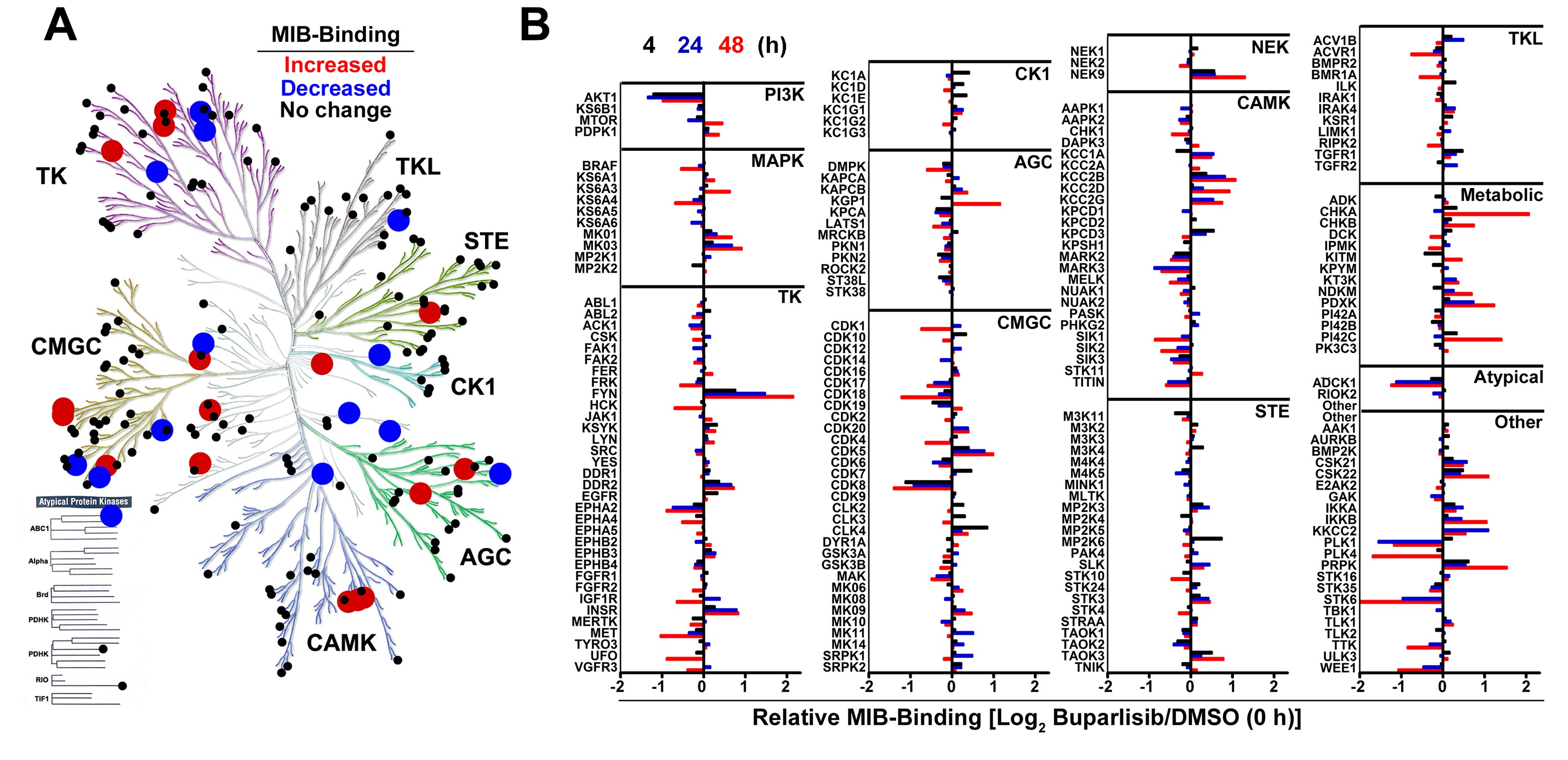
We previously used MIB-MS to show that adaptive kinome reprogramming promotes single agent kinase inhibitor resistance.27,28 We thus used this technique to examine the adaptive kinome response of TRP astrocytes to the PI3Ki buparlisib. Buparlisib induced widespread kinome changes, with alterations in multiple kinase families (Supplementary Fig. S4A). Kinome response was dynamic and the patterns of activity differed among kinases (Fig. 2A, Supplementary Fig. S4B, Supplementary Table S3). Three major temporal patterns were discernible, as we have previously described (Fig. 2B).27,28 Buparlisib induced sustained inhibition of proximal (eg, Akt1, pattern 1) and transient inhibition of distal PI3K (eg, mTor), followed by reactivation (pattern 2). Activation of alternative pathways such as MAPK (eg, extracellular signal-regulated kinase [ERK]1/2) was also apparent (pattern 3).
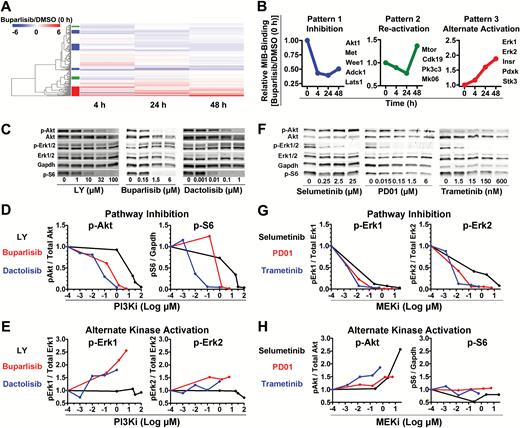
Single agent PI3K/MEKi induces dynamic kinome changes in TRP astrocytes. (A) A heatmap demonstrated temporal changes 4–48 h after buparlisib. (B) Dynamics of select kinases illustrated 3 response types: sustained inhibition (pattern 1, blue), reactivation (pattern 2, green), and alternate pathway activation (pattern 3, red). Graphs show the first kinase listed. (C) Representative immunoblots of PI3Ki-treated TRP astrocytes at 24 h showed that potency was directly associated with dose-dependent decreases of proximal (pAkt) and distal (pS6) PI3K (D), while the more potent PI3Ki, buparlisib and dactolisib, induced alternate MAPK (pERK1/2) activation (E). (F) Immunoblots performed on TRP astrocytes 24 h after selumetinib, PD01, or trametinib showed that MEKi potency directly associates with MAPK inhibition (G) and alternate activation of proximal, but not distal, PI3K signaling (H). A representative immunoblot quantification is shown (N = 1–5 biologic replicates, mean = 2.5).
Immunoblots of TRP astrocytes treated with buparlisib for 4 (data not shown) and 24 h (Fig. 2C–E) showed that proximal (pAkt) and distal (pS6) PI3K signaling were inhibited at 4 h and sustained for 24 h, while MAPK signaling progressively increased over 24 h. Next we assessed how potency influenced PI3K inhibition and alternate MAPK activation. All PI3Ki caused dose-dependent inhibition of proximal and distal PI3K signaling, and inhibition was directly associated with drug potency (Fig. 2C, D). Moreover, both of the more potent PI3Ki, buparlisib and dactolisib, induced alternate MAPK activation (Fig. 2C, E). Taken together, these results suggested that alternate MAPK activation contributes to PI3Ki resistance.
MEKi Induces MAPK Inhibition and Alternate PI3K Activation In Vitro
We found that PI3Ki induced MAPK activation (Fig. 2B–E) and that multiple MEKi reduced TRP astrocyte growth (Fig. 1C, Supplementary Figures S1C, S2B). MEKi have been shown to induce alternate PI3K activation in multiple cancer types.32–34 We therefore performed immunoblots on TRP astrocytes 24 h after treatment with increasingly potent MEKi to determine their effects on MAPK and PI3K signaling (Fig. 2F). All 3 MEKi caused dose-dependent decreases in MAPK signaling, and dose/potency was directly associated with degree of inhibition (Fig. 2F, G). Activation of proximal, but not distal, PI3K signaling was evident at 24 h with all 3 MEKi, and activation was associated directly with dose/potency (Fig. 2F, H).
A Subset of GBM and PDX Models Have Hyperactive Kinomes Featuring MAPK Activation
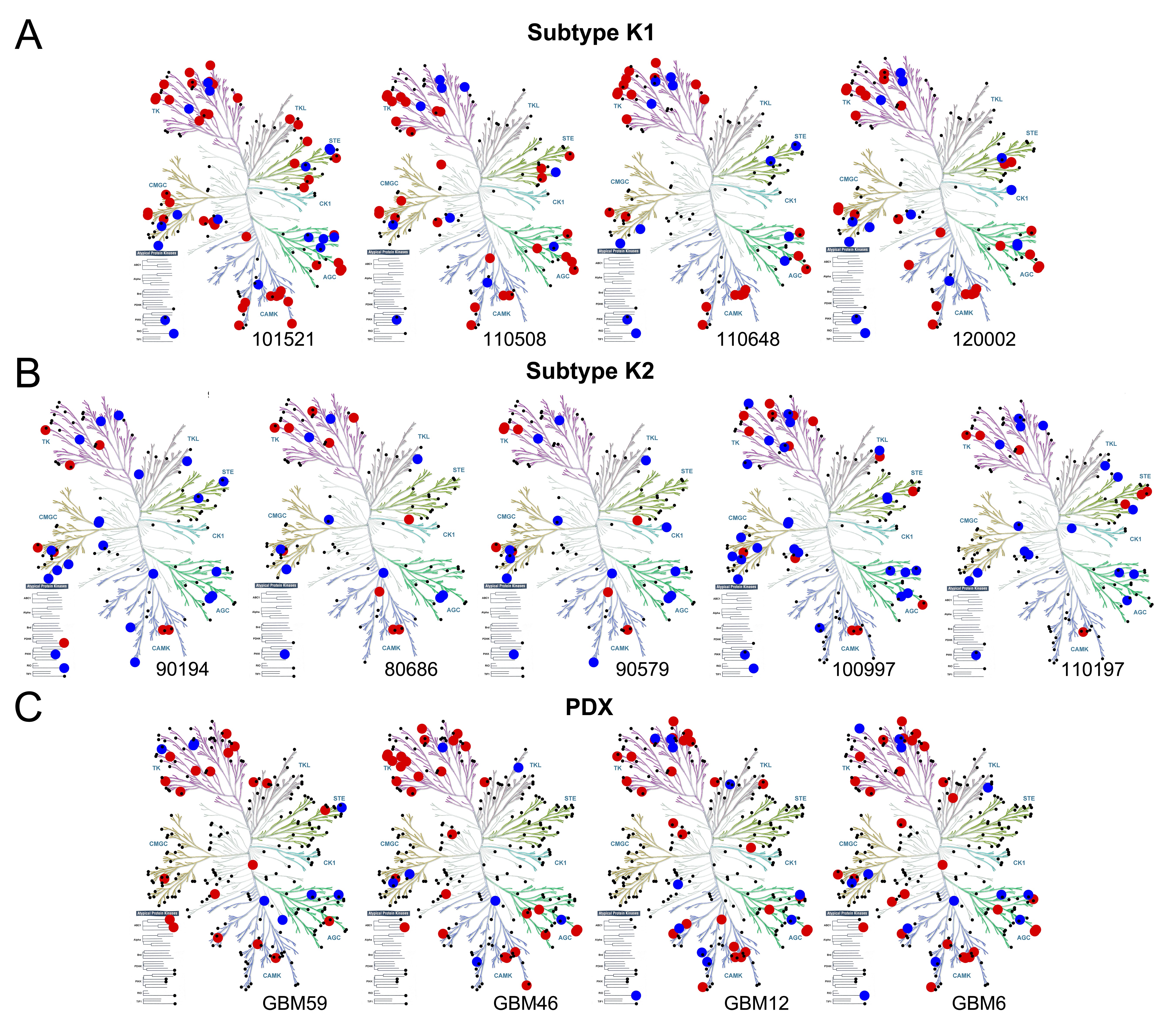
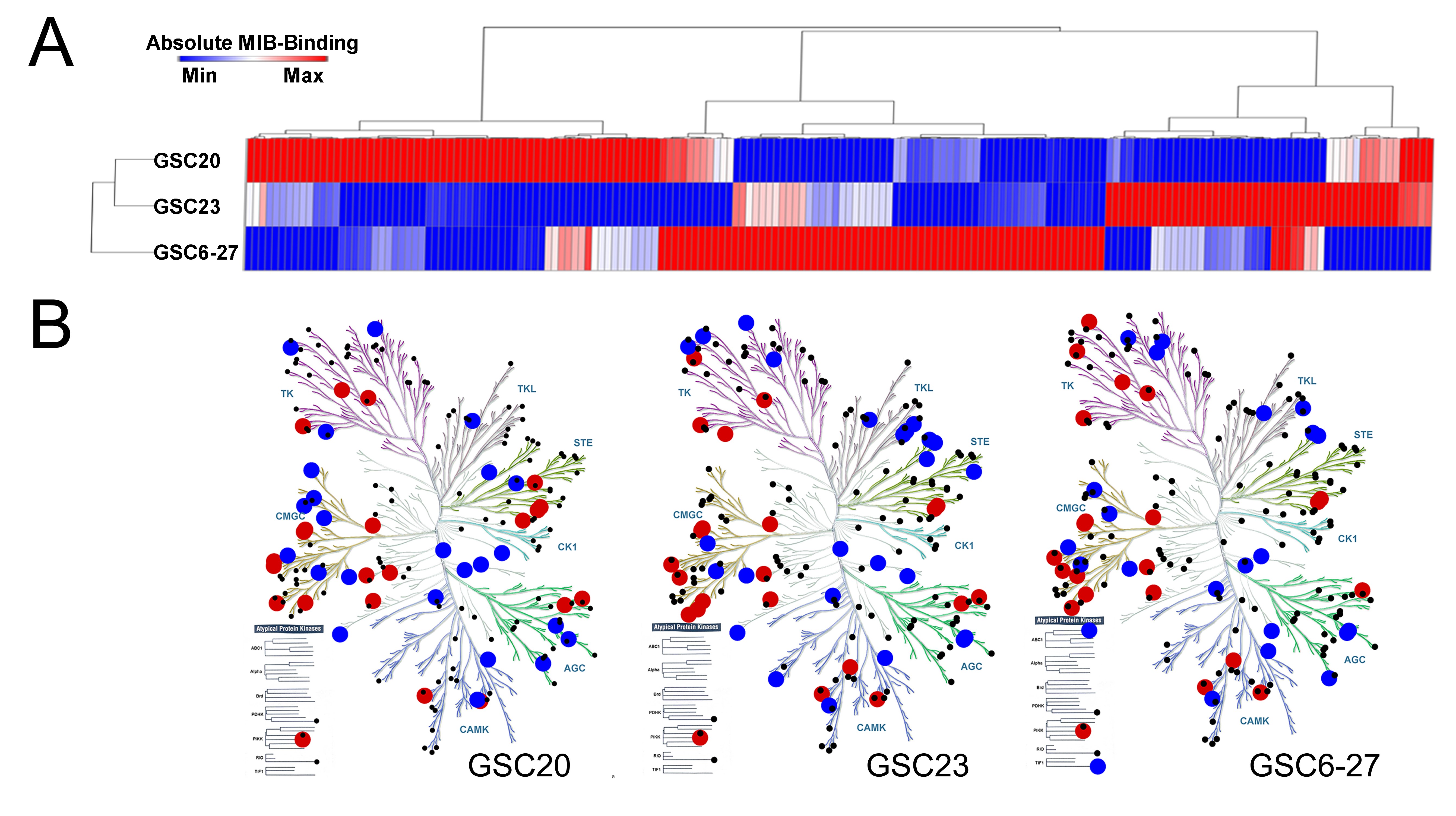
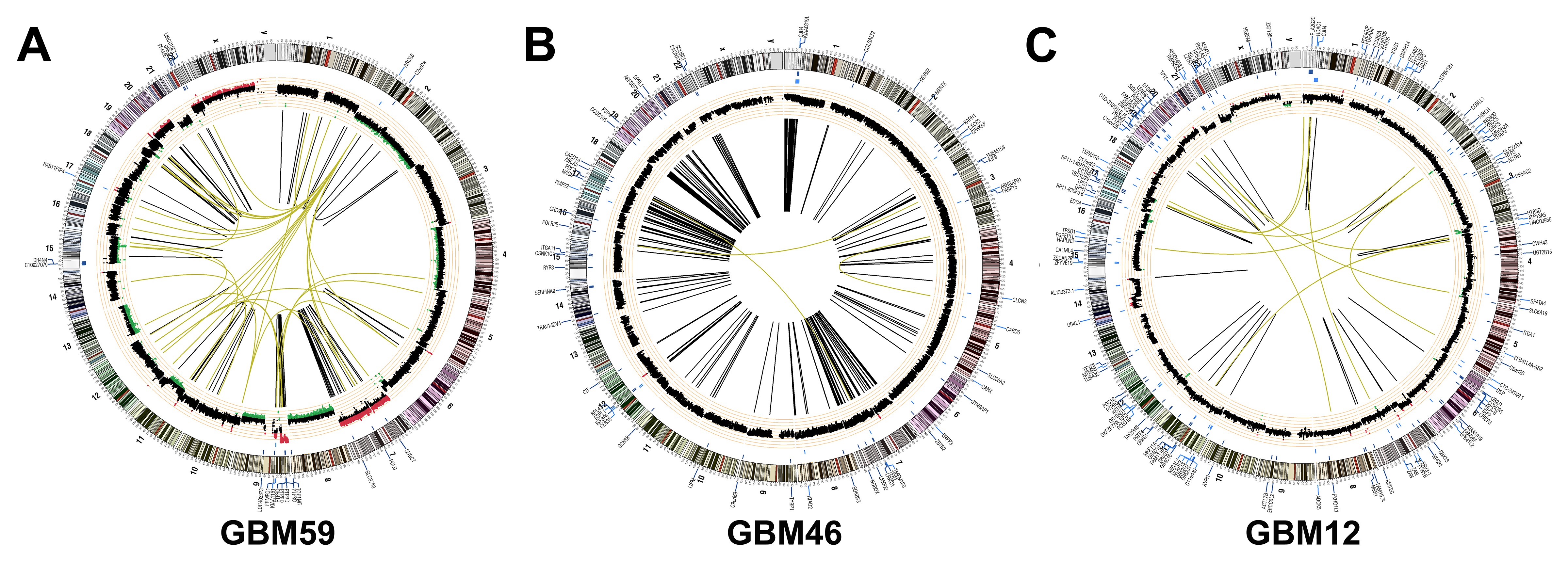
GBM has extensive genome and transcriptome heterogeneity.3,4 However, details on the basal activation state of its kinome are limited. We therefore analyzed baseline kinome profiles of human GBM-derived ECL (Fig. 3A) and patient samples (Fig. 3B) via MIB-MS. Hierarchical clustering showed kinome heterogeneity, with relative hyperactivation of unique kinases in each model and patient sample. Principal components analysis stratified patient tumors into 2 kinome subtypes (K1 and K2) (Fig. 3C). Heatmaps (Fig. 3B) and kinome trees (Fig. 3D, E, Supplementary Fig. S5A, B) showed that K1 tumors had a relatively hyperactive kinome compared with K2. Among the hyperactivated K1 kinases were MAP2K1 (MEK1), RPS6KA2 (ribosomal S6 kinase [RSK]3), MAPK3 (ERK2), and MAPK1 (ERK1) (Fig. 3F), suggesting that MAPK hyperactivation may be an attractive therapeutic target in these tumors. Baseline kinome profiles of subcutaneous (Fig. 3G, H, Supplementary Fig. S5C) and cultured (Supplementary Fig. S6A) PDX were also variable. GSC23 and GSC6-27 harbored activation of numerous kinases within the tyrosine kinase (TK) and CMGC families, similar to the K1 subtype of patient samples (Supplementary Fig. S5A, S6B). Moreover, the PDX GBM59, GBM46, and GBM12 had heterogeneous genomes (Supplementary Fig. S7), but all harbored activation of numerous TKs (Supplementary Fig. S5C, Supplementary Table S4).
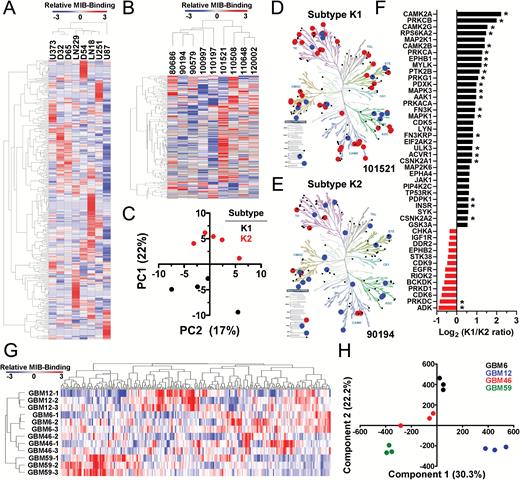
GBMs have heterogeneous kinomes. Heterogeneous kinome activity was evident in (A) ECL and (B) tumor samples from human GBM patients. (C) Principal components analysis showed 2 kinome subtypes of human tumors (K1, K2). (D) K1 had hyperactivation relative to K2 (E) tumors (Supplementary Figure S5A, B). Kinases with ≥2x (red) or ≤0.4x (blue) relative MIB binding are indicated; other detected kinases (black). A waterfall plot shows the most differentially activated kinases (F). Kinases significantly (P < 0.05) enriched in K1 (black) and K2 (red) are indicated (*). Heterogeneous kinome activity was also evident in subcutaneous GBM PDX (G, Supplementary Figure S5C). Principal components analysis demonstrated that although variable, biologic replicates of subcutaneous GBM PDX were more similar to each other than to different PDX models (H).
PI3Ki and MEKi Are Synergistic In Vitro
Data from Fig. 2 suggest that PI3K and MAPK are reciprocal bypass pathways that promote cell survival when either pathway alone is inhibited. We therefore assessed whether dual PI3K/MEKi caused inhibition of both pathways. Immunoblots of TRP astrocytes 24 h after buparlisib plus selumetinib (Fig. 4A) or trametinib (Fig. 4B) showed a dose-dependent inhibition of both PI3K/MAPK signaling. The concentration of buparlisib required for PI3K inhibition was similar regardless of the MEKi used, but MEKi potency was directly associated with MAPK inhibition.
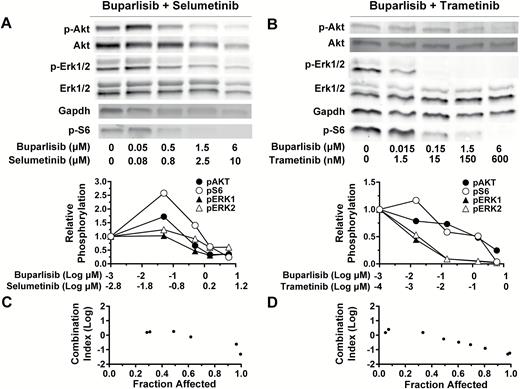
PI3K/MEKi are synergistic, and dual therapy inhibits alternate pathway activation. Immunoblots of TRP astrocytes treated with buparlisib and either selumetinib (A) or trametinib (B) for 24 h showed that dual treatment blocks both PI3K and MAPK signaling. Buparlisib and selumetinib were synergistic in TRP astrocytes at 2–50 and 3.3–83 µM (≥0.6 fraction affected) (C). Buparlisib and trametinib were synergistic in TRP astrocytes at 0.75–50 and 0.08–5 µM (≥0.5 fraction affected) (D). A representative immunoblot quantification is shown (N = 1–4 biologic replicates, mean = 2.5).
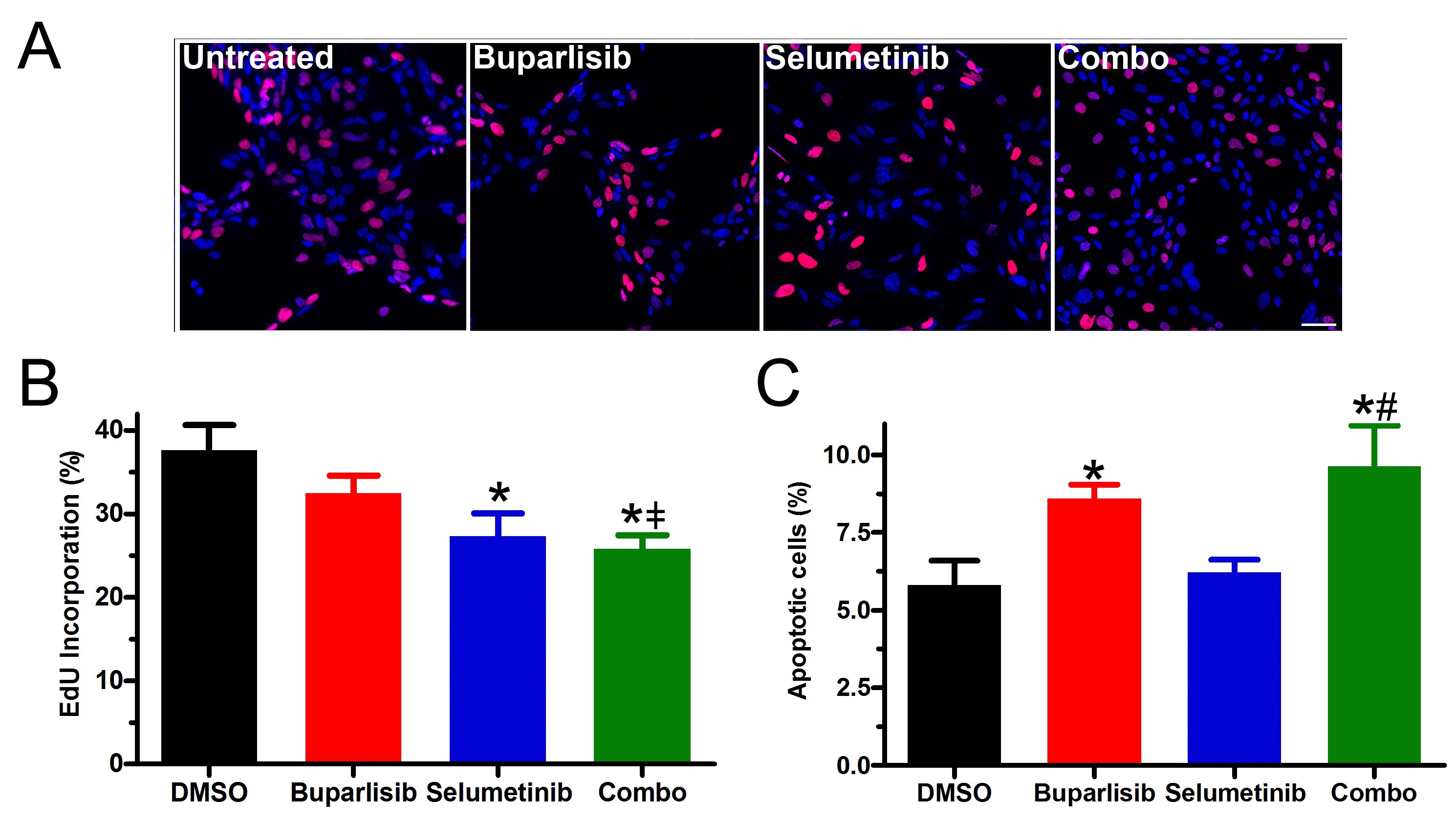
Next, we assessed whether PI3K/MEKi functioned synergistically. Buparlisib/selumetinib inhibited growth and was synergistic in TRP astrocytes (Fig. 4C), likely due to the combinatorial effects of selumetinib-induced decreases in proliferation (Supplementary Fig. S8A, B) and buparlisib-induced increases in apoptosis (Supplementary Fig. S8C). Buparlisib plus the more potent MEKi trametinib also inhibited growth and was synergistic (Fig. 4D). However, PI3K/MEKi concentrations required for synergism were dramatically reduced with buparlisib/trametinib, compared with buparlisib/selumetinib.
PI3Ki and MEKi Are Effective in Subcutaneous TRP Allografts
The blood–brain barrier can hinder drug penetrance and limit efficacy.13 Dactolisib and trametinib, but not buparlisib and selumetinib, have poor brain penetrance.35–37 We therefore first tested PI3K/MEKi efficacy in subcutaneous TRP allografts to eliminate the impact of variable brain penetrance. Single agents were used at previously described doses (Supplementary Table S2).35,36,38 Both PI3Ki reduced tumor growth (Supplementary Fig. S9A, B) and inhibited PI3K (pAkt) signaling by ~50% (Supplementary Fig. S10A, B), but neither extended survival (Supplementary Fig.S9C). Both MEKi also delayed tumor growth (Supplementary Fig. S9D, E) and decreased MAPK (pERK) signaling >60% (Supplementary Fig. S10A, C). Trametinib, but not selumetinib, extended survival (Supplementary Fig. S9F).
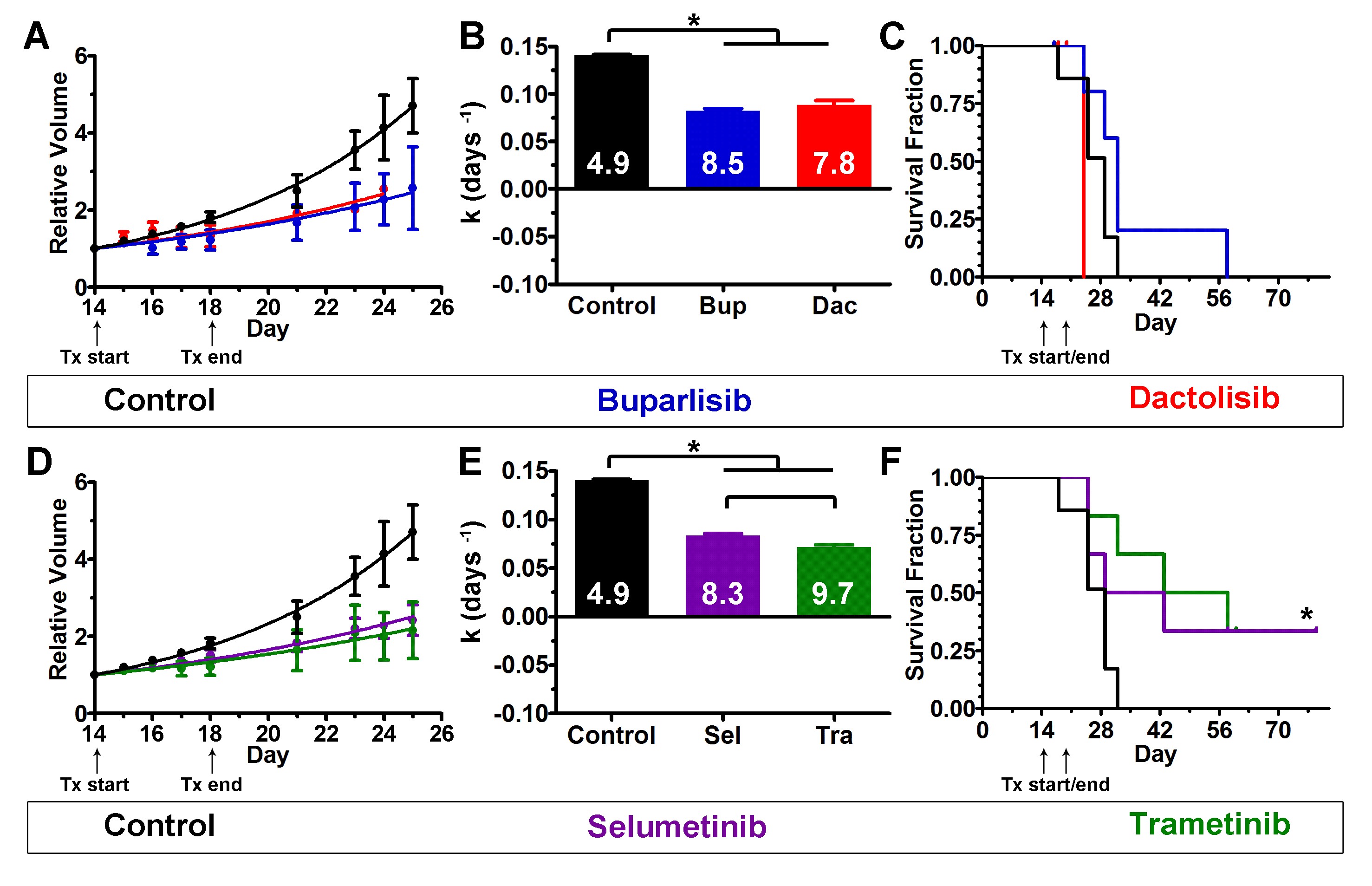
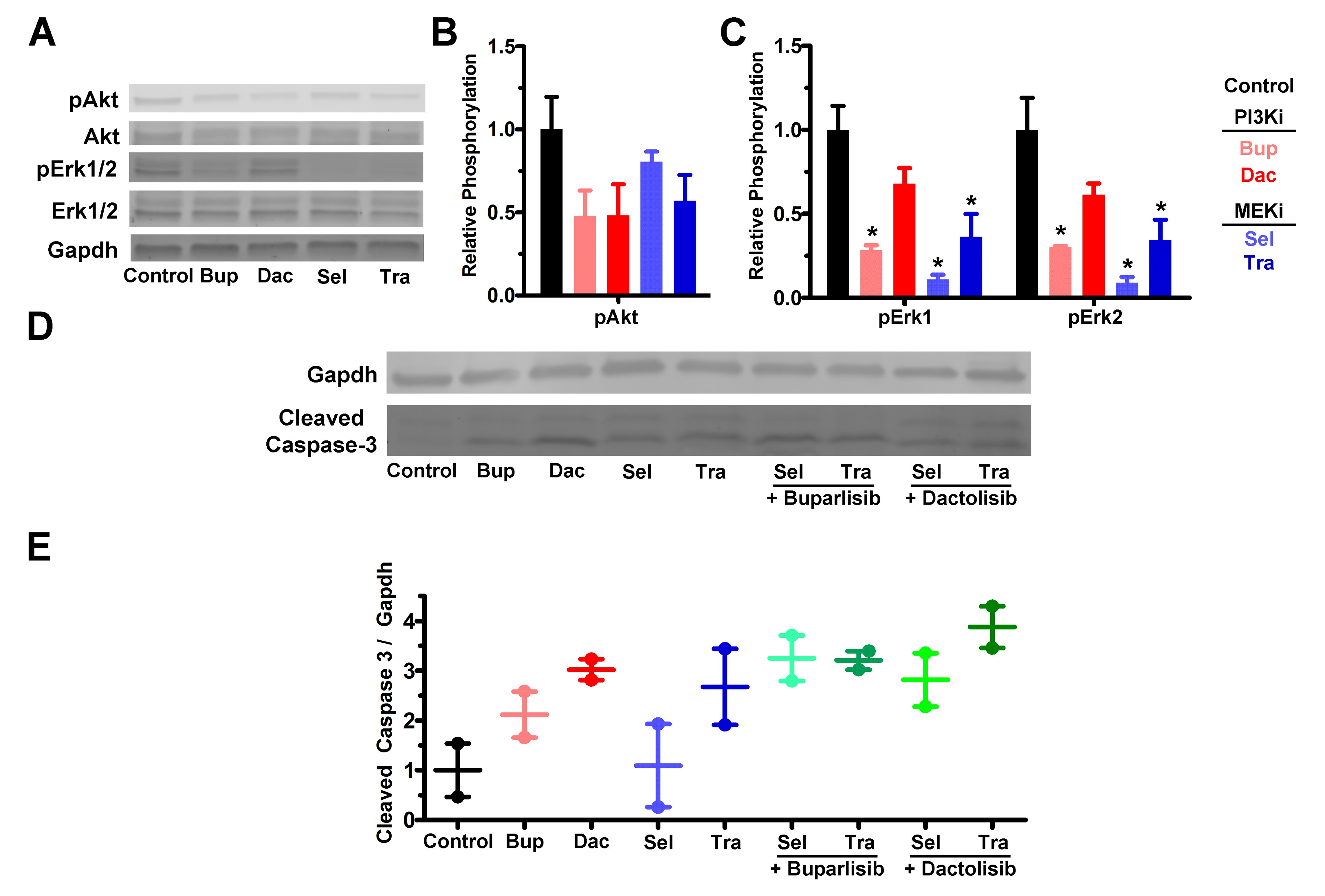
Because PI3K/MEKi were synergistic in vitro (Fig. 4), we hypothesized that combinations would be more effective than single agents in vivo. However, drug-induced toxicity can be amplified when combinations are used, necessitating dose reduction. We found that the maximum tolerated dose of buparlisib/selumetinib was achieved at 1.2- to 2.1-fold lower doses in non–tumor bearing mice (Supplementary Table S2, data not shown). This combination was well tolerated and effective in an intracranial xenograft model of triple-negative breast cancer when used on a 5 days on/2 days off schedule.37 Dactolisib/selumetinib doses were based on published literature.38 For in vivo experiments with the PI3Ki buparlisib or dactolisib in combination with MEKi trametinib, we empirically kept the PI3Ki doses constant and empirically halved the trametinib dose, as we did with selumetinib (Supplementary Table S2). Combination treatments, as well as most single agents, caused an increase in cleaved caspase-3, an indicator of apoptosis (Supplementary Fig. S10D, E). Buparlisib/selumetinib caused tumor regression (Fig. 5A, B) and increased survival (Fig. 5C). Buparlisib plus the more potent MEKi trametinib also significantly reduced growth (Fig. 5D, E) and extended survival (Fig. 5F). Both combinations were more effective than either drug alone.
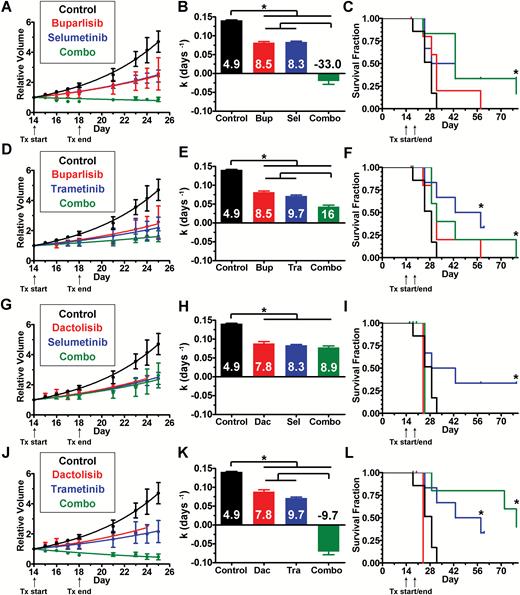
PI3Ki and MEKi treatment inhibited growth of subcutaneous TRP tumors. Buparlisib plus either selumetinib (A–C) or the more potent MEKi trametinib (D–F) delayed tumor growth more than either drug alone and improved survival compared with controls (*P ≤ 0.01). The more potent PI3Ki dactolisib plus either selumetinib (GH) or the more potent MEKi trametinib (JK) delayed tumor growth compared with controls (*P < 0.0001). Dactolisib/trametinib delayed growth more than either treatment alone (*P < 0.0001). Dactolisib/selumetinib did not improve survival compared with controls (I), but dactolisib/trametinib did (L) (*P = 0.01). Doubling times (days) are indicated (BEHK).
Similar results were obtained with the more potent PI3Ki dactolisib combined with MEKi. Dactolisib/selumetinib significantly reduced tumor growth (Fig. 5G, H) but did not extend survival (Fig. 5I). However, mice treated with dactolisib ± selumetinib, but not the more potent dactolisib/trametinib combination, exhibited drug-induced toxicity, particularly rapid onset of lethargy. Dactolisib/trametinib-induced tumor regression (Fig. 5J, K), increased survival (Fig. 5L) and was more effective at growth inhibition than either drug alone. These results indicate that combination therapy with PI3K/MEKi is effective in subcutaneous TRP allografts, with the more potent inhibitors being particularly effective.
Selumetinib Delays Orthotopic TRP Allograft Growth
We found that TRP astrocytes were sensitive to single agent PI3K/MEKi (Fig. 1, Supplementary Fig. S2) and that dual treatment was synergistic (Fig. 4C, D) in vitro. Moreover, single agents delayed subcutaneous tumor growth, but dual treatment was more effective (Fig. 5). We therefore hypothesized that these treatments might be effective in orthotopic TRP allografts. We therefore tested the brain-penetrant PI3Ki buparlisib and/or MEKi selumetinib in this model and found that selumetinib ± buparlisib transiently delayed tumor growth, but buparlisib alone did not (Fig. 6A, B). Selumetinib modestly but significantly prolonged survival, but all mice eventually succumbed to recurrent disease. Buparlisib ± selumetinib failed to extend survival (Fig. 6C).
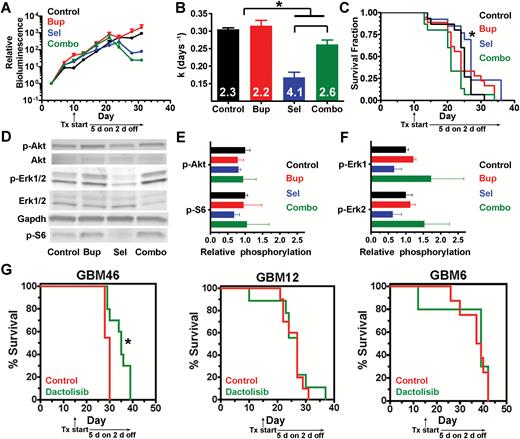
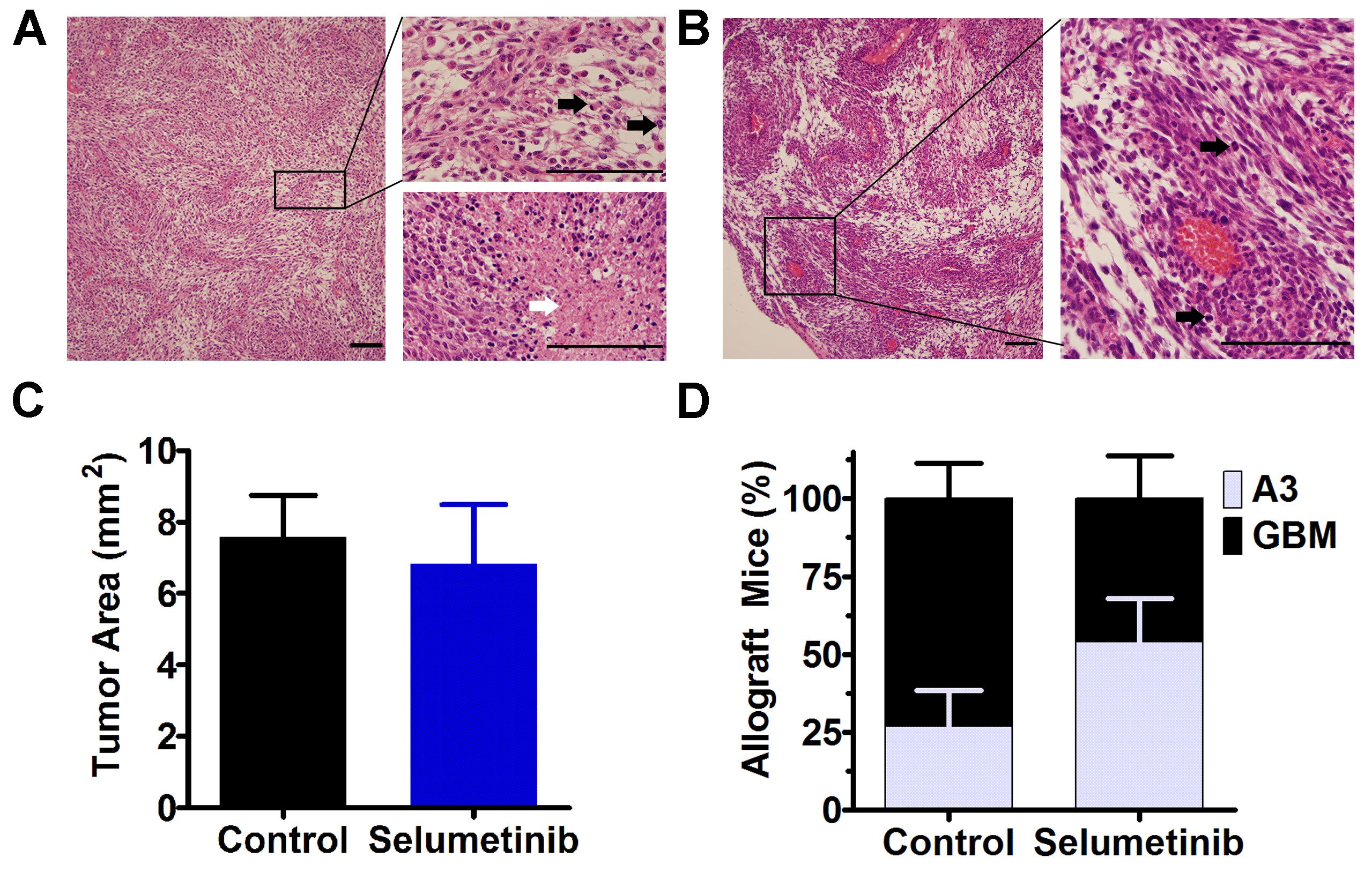
Response of orthotopic TRP allografts and PDX to PI3Ki and MEKi. In TRP allografts (A–F), selumetinib ± buparlisib delayed growth (*P ≤ 0.03) (AB). Doubling times (days) are indicated (B). Selumetinib alone improved survival (*P = 0.03) (C). Representative immunoblot (D) and quantification showed no change in PI3K (E), but selumetinib trended towards decreased MAPK signaling (P = 0.06) (N = 3 biologic replicates) (F). In GBM PDX, dactolisib modestly improved GBM46 (*P = 0.003), but not GBM12 and GBM6 survival (G).
Next, we defined the pharmacodynamic effects of treatment on PI3K/MAPK signaling in vivo by performing immunoblots on tumors harvested from terminal mice (Fig. 6D). Buparlisib did not affect PI3K signaling (Fig. 6E), either alone or in combination with selumetinib. In contrast, selumetinib caused a modest MAPK decrease (Fig. 6F) when used alone, but not in combination with buparlisib, likely due to the ~2-fold increased dose. Because selumetinib was the most effective treatment, we next compared the histopathology of terminal control and selumetinib-treated mice (Supplementary Fig. S11A, B). Tumor area was similar between groups (Supplementary Fig. S11C), but progression to GBM was more frequent in untreated tumors (Supplementary Fig. S11D). Taken together, these data suggest that selumetinib was effective due to inhibition of MAPK (~50%, Fig. 6F) with minimal PI3K reactivation when used alone at ~2-fold higher dose. Moreover, they suggest that buparlisib alone was ineffective due to minimal PI3K inhibition despite favorable CNS pharmacokinetics. Finally, these data suggest that the reduced doses of both drugs required for combination therapy limited target inhibition as well as efficacy in orthotopic GBM models.
GBM PDX Response to Dactolisib Is Heterogeneous
Human GBM PDX are valuable models for preclinical drug development because they faithfully recapitulate the genotypic and phenotypic characteristics of their parent tumors.39 Although dactolisib was the most effective PI3Ki in cultured TRP astrocytes and PDX models (Figure 1A, B), its effectiveness in a preclinical GBM model was limited.35 To confirm this finding, we examined dactolisib efficacy in a panel of orthotopic PDX models. Survival of mice harboring GBM46, but not GBM12 or GBM6, was extended by dactolisib, but its effects were modest (17% increased survival, Fig. 6G). This heterogeneous response of PDX to dactolisib highlights the importance of identifying predictive biomarkers for the development of kinase inhibitors.
Discussion
PI3K/MAPK Mutations Influence Targeted Inhibitor Efficacy
GBM are molecularly heterogeneous, but the vast majority harbor activated MAPK and PI3K.3,8 Core pathway mutations influence single agent kinase inhibitor efficacy in GBM ECL models.4,32,40 However, preclinical drug studies with ECL correlate poorly with clinical outcomes.13 PDX and nGEM are more genetically faithful and thus may be more predictive.41 We found that cultured PDX and TRP nGEM were sensitive to PI3Ki or MEKi, with increased potency directly associated with efficacy (Fig. 1, Supplementary Figures S1–S3).
Dual PI3K/MEKi therapies have shown efficacy in some preclinical GBM models and have demonstrated the importance of PI3K/MAPK crosstalk in GSC maintenance.32,34,35 We have shown that both pathways are critical for astrocyte de-differentiation into GSCs in TRP nGEM models.16,20 We expanded these results here by determining that the PI3K/MEKi concentrations required for synergism in TRP astrocytes were dramatically reduced when a more potent MEKi was used (Fig. 4C, D).
Bypass Pathways Promote Resistance to Single Agent Kinase Inhibitors
We and others have shown that PI3K/MAPK crosstalk is essential for GSC genesis, maintenance, and tumorigenicity, suggesting that dual inhibition of these pathways may be effective.16,20,34 Additional evidence for the necessity of combination treatments comes from our own preclinical work in breast cancer, showing that dynamic kinome reprogramming promotes single agent kinase inhibitor resistance.27,28 We extended this work to GBM here and found that buparlisib induces extensive kinome reprogramming in TRP astrocytes (Fig. 2). Although we focused primarily on alternate MAPK activation, dynamic kinome profiling showed that multiple pathways were altered in response to PI3Ki and may thus promote resistance. Future work will be required to investigate these pathways.
We and others have found that PI3K and MAPK are reciprocal bypass pathways that promote resistance to drugs targeting either pathway alone (Fig. 2 and 4).32,34,35 However, these dynamic responses may be influenced by mutational activation of PI3K/MAPK signaling as well as by different mutations or tumor cells of origin. Here we found that sensitivity of PDX to MEKi or PI3Ki was variable (Fig. 1 and 6, Supplementary Fig. S1), but the reason for these differences was not readily apparent based on their genomic or baseline kinome profiles (Fig. 3, Supplementary Fig. S7, Supplementary Table S4). Systematic chemovulnerability profiling of a large, integrated panel of PDX and nGEM with diverse mutations and cells of origin may be necessary to further elucidate the contours of GBM kinome dynamics and aid in the development of combination treatments tailored to specific tumor subsets.
We previously showed that genomic heterogeneity in breast cancer extends to its kinome.27,42 Here we extend these findings by demonstrating that MIB-MS analysis can stratify human GBM samples and PDX/ECL models based on their kinome profiles (Fig. 3). Based on our experience with an ongoing “window” trial of neoadjuvant kinase inhibitor therapy in breast cancer, we anticipate that MIB-MS–based kinome profiling of pre- and posttreated GBM patient samples will ultimately result in identification of novel resistance mechanisms and facilitate design of rational combination treatments.42
Drug Potency Influences Single and Dual Agent Efficacy
Increased potency facilitates target modulation at lower drug concentrations and dose reduction in vivo. We found that potent PI3K/MEKi enhanced growth inhibition and synergism in vitro (Fig. 1 and 4, Supplementary Figures S1–S3). Dactolisib and trametinib have poor brain pharmacokinetics. Use of orthotopic but not subcutaneous TRP models would therefore preclude comparison of target inhibition and efficacy in vivo to their brain-penetrant counterparts, buparlisib and selumetinib. Single agent PI3Ki buparlisib and dactolisib were equally effective in subcutaneous TRP allografts, but trametinib was more effective than the less potent MEKi selumetinib (Supplementary Fig. S9). Three of 4 dual treatments were more effective than their corresponding single agents, particularly the most potent combination, buparlisib/trametinib (Fig. 5). However, dactolisib ± selumetinib induced systemic toxicity, likely limiting their effectiveness.
We investigated efficacy of the brain-penetrant PI3K/MEKi combination, buparlisib/selumetinib, in orthotopic TRP allografts. Selumetinib alone caused signaling inhibition and was most effective. Lack of buparlisib/selumetinib efficacy was likely due to dose reduction (~2-fold for selumetinib) required to prevent toxicity (Fig. 6, Supplementary Table S2), consistent with dose limiting toxicity for kinase inhibitor combinations found in clinical settings.14 We also tested selumetinib/buparlisib in intracranial triple-negative breast cancer xenografts and found that target inhibition occurred in sensitive but not resistant models.43 This suggests that drug levels within orthotopic TRP allografts were insufficient to cause signaling inhibition and affect outcomes; this limitation may be overcome using alternative delivery approaches (eg, nanoparticles) that improve brain penetrance and reduce systemic toxicity.44
Conclusion
Monitoring kinome dynamics by MIB-MS represents a valuable tool to identify bypass pathways and design rational drug combinations. Its use in directing preclinical trials in genetically faithful models, such as the nGEM model used here, can aid in drug and predictive biomarker development. Our results suggest that highly potent, brain-penetrant kinase inhibitor combinations that target resistance pathways will likely be required to design effective clinical trials in molecularly defined GBM patients.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
C.R.M. was a Damon Runyon–Genentech Clinical Investigator supported by the Damon Runyon Cancer Research Foundation (CI-45-09), Department of Defense (W81XWH-09-2-0042), UNC University Cancer Research Fund (UCRF), and NIH National Center for Advancing Translational Sciences (550KR21202). R.S.M. is a Robert H. Wagner Scholar. R.S.M. and D.M.I. were supported by the UNC/Howard Hughes Medical Institute Graduate Training Program in Translational Medicine. C.R.M. (R01CA204136), D.M.I. (T32GM007092; T32CA071341), and G.L.J. (R01GM101141) were supported by NIH. M.J.H. was supported by the American Brain Tumor Association. The Translational Pathology Laboratory (TPL) and Small Animal Imaging Facility (SAIF) were supported by P30CA016086 and UCRF. TPL was supported by P30ES010126 and W81XWH-09-2-0042. M.E.B. and H.D.D. were supported by the Ben & Catherine Ivy Foundation.
Acknowledgments
We thank G. Yancey Gillespie for providing cell lines and UNC TPL and SAIF for histology and animal imaging assistance.
Conflict of interest statement. Gary L. Johnson and Lee M. Graves are co-founders of KinoDyn. Other authors declare no conflicts of interest.
References



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}