




Abstract
Ewing's family tumors (EFTs) are highly malignant tumors arising from bone and soft tissues that exhibit EWS–FLI1 or variant EWS–ETS gene fusions in more than 85% of the cases. Here we show that CIC, a human homolog of Drosophilacapicua which encodes a high mobility group box transcription factor, is fused to a double homeodomain gene DUX4 as a result of a recurrent chromosomal translocation t(4;19)(q35;q13). This translocation was seen in two cases of soft tissue sarcoma diagnosed as Ewing-like sarcoma. CIC–DUX4 exhibits a transforming potential for NIH 3T3 fibroblasts, and as a consequence of fusion with a C-terminal fragment of DUX4, CIC acquires an enhanced transcriptional activity, suggesting that expression of its downstream targets might be deregulated. Gene expression analysis identified the ETS family genes, ERM/ETV5 and ETV1, as potential targets for the gene product of CIC–DUX4. Indeed, CIC–DUX4 directly binds the ERM promoter by recognizing a novel target sequence and significantly up-regulates its expression. This study clarifies the function of CIC and its role in tumorigenesis, as well as the importance of the PEA3 subclass of ETS family proteins in the development of EFTs arising through mechanisms different from those involving EWS–ETS chimeras. Moreover, the study identifies the role of DUX4 that is closely linked to facioscapulohumeral muscular dystrophy in transcriptional regulation.
INTRODUCTION
Recurrent chromosomal translocations are observed frequently among bone and soft tissue tumors in a tumor type-specific manner (1). Many of these chromosomal translocations involve genes encoding transcription factors (2), and fusion to the partner gene on a different chromosome results in the acquisition of novel functions. These oncogenic transcription factors may dysfunctionally regulate their downstream targets, thus making an important contribution to the multi-step biological process of carcinogenesis. In bone and soft tissue tumors, there is strict relationship between tumor types and gene fusions, suggesting that a chimeric gene may be generated in multi-potent stem cells and may determine a tumor phenotype by modulating its differentiation process. Alternatively, a tumorigenic potency of a chimeric gene may be effective only in a certain cell type.
Ewing family tumors (EFTs) include Ewing's sarcoma, Askin's tumor and peripheral primitive neuroectodermal tumors and they afflict children and young adults (3). EFTs are aggressive tumors with massive and destructive growth at the primary loci, and they have a high incidence of distant metastases, with eventual relapse in ~50% of cases even after 5 years, despite extensive treatment (4). Most cases of EFTs are characterized cytogenetically by fusion between the 5′ site of the EWS gene at 22q12 and the 3′ site of the members of the ETS family of transcription factors including FLI1 (11q24), ERG (21q22), ETV1 (7p22), E1AF (17q12) or FEV (2q33) (5–9). In 85% of the cases, EWS is fused to FLI1 and the chimeric EWS–FLI1 protein is composed of the N-terminal transactivation domain of EWS and the C-terminal ETS DNA-binding domain. It is presumed that an altered pattern of gene expression results and that this is of importance in tumorigenesis (10–13). In addition, EWS as a TET family protein associates with RNA splicing factors, and EWS–FLI1 modulates RNA splicing (14). It remains to be clarified whether the EWS–ETS fusion gene is essential for carcinogenesis of EFTs, as EWS–ETS expression is not sufficient to induce fully developed EFTs (15). A FUS–ERG fusion has been identified in a few cases of Ewing's sarcoma (16). In these cases, FUS serves a function equivalent to that of EWS. However, it is also possible that unknown oncogenes exist that are structurally unrelated but that function in the same molecular pathway as EWS–ETS in oncogenesis of EFTs. Alternatively, there may be a novel disease category that is related to but biologically not identical with EFTs.
We describe here two cases of Ewing-like sarcomas containing a novel chromosomal translocation t(4;19)(q35;q13). These tumors shared pathologically similar features with Ewing's sarcoma. However, apparent lack of EWS–ETS fusion and weak expression of CD99/MIC2, a marker of EFTs, made diagnosis and classification of these tumors difficult. CIC, a human homolog of the Drosophilacapicua gene, was identified at the 19q13 breakpoint, where it was found fused to the DUX4 double homeodomain gene. Furthermore, CIC–DUX4 functions as a transcriptional activator and two members of the PEA3 subfamily genes are its direct targets. These studies illuminate a molecular network of transcription factors and demonstrate the important role of ETS family proteins in development of mesenchymal tumors and phenotypes for small round cell tumors.
RESULTS
Pathological examination and chromosome analysis
Two cases of malignant soft part tumors showed the typical histopathological features of Ewing's sarcoma display t(4;19)(q35;q13). Details of the clinicopathological features of these two cases are shown in Materials and Methods. As shown in Figure 1, characteristic features were diffuse infiltrative growth of small round tumor cells (Fig. 1A) that were positive for glycogen granules within their cytoplasms (Fig. 1B). Immunohistochemical staining of the tumor cells revealed that they expressed CD99/MIC2 (Fig. 1C), though the expression is much weaker than that of typical EFT (Fig. 1D).
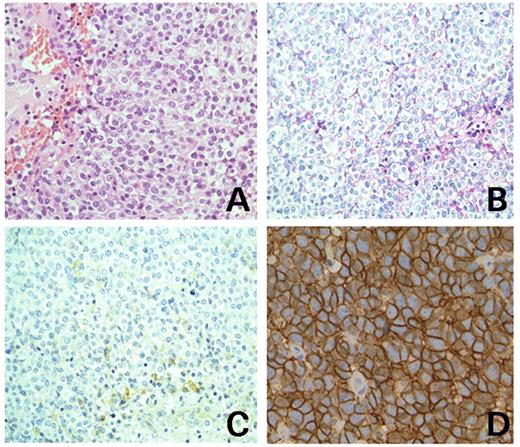
Microscopic features of the Ewing's sarcoma with t(4;19)(q35;q13). (A) The hematoxylin and eosin-stained section shows diffuse proliferation of small round tumor cells (original magnification, 20×). (B) Periodic acid Schiff's staining. The small number of glycogen granules in the cytoplasm are shown (original magnification 20×). (C) Immunohistochemical staining shows that CD99/MIC2 is weakly positive for tumor cells (original magnification, 20×). (D) CD99/MIC2 staining of typical EFT (original magnification, 40×).
Conventional chromosome analysis revealed that both cases carried reciprocal t(4;19)(q35;q13) as sole translocations and no chromosome 22 rearrangements were observed (data not shown). Dual color fluorescence in situ hybridization (FISH) for 19q13 showed that the breakpoint was located within the bacterial artificial chromosome (BAC) clone RP11-569M1 (Fig. 2). RP11-569M1 covers the 230 kb region within 19q13 (Fig. 2C, top) and contains at least 10 genes. Using multiple probes and tumor DNA samples, the breakpoint was determined by Southern blotting. DNA rearrangements were detected in both cases when a probe derived from exon 10 of the CIC gene encoding a high mobility group (HMG) box containing protein was used (Fig. 2C and D), indicating that CIC is a target for the 19q13 break.
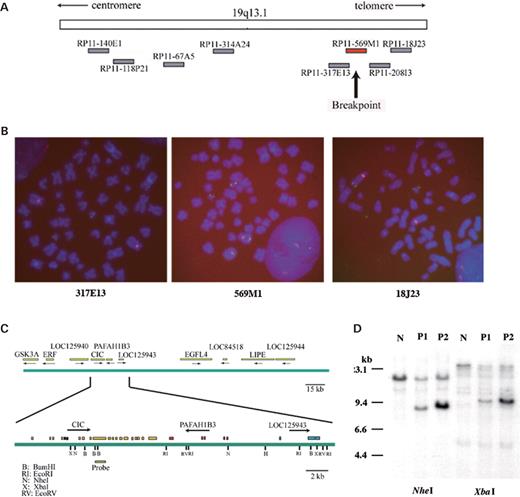
Cloning of the chromosome 19 breakpoint. (A) A physical map of 19q13.11–13.31 and BAC clones used in the FISH analysis. (B) FISH analysis of sarcoma cells. The green fluorescence indicates probes for the 19q13 region. RP11-317E13 (left), RP11-569M1 probe (center) and RP11-18J23 (right). The red fluorescence indicates the centromeric region of chromosome 4. A split of one copy of the RP11-569M1 is shown both in metaphase and in interphase nuclei. (C) The fine map of RP11-569M1. The arrow indicates the transcriptional orientation of each gene (top). The structure of the CIC gene and the location of the probe used for Southern blotting are indicated (bottom). (D) Southern blotting. Rearranged bands were detected in both cases by NheI or XbaI digestion. N, normal control; P1, case 1; P2, case 2.
Detection of the CIC–DUX4 chimera
A subgenomic library was constructed by using DNA obtained from the tumor cells derived from case 2. A 5.0 kb BamHI fragment containing both 19q and 4q regions was isolated, and sequence analysis revealed that the genetic breakpoint at 19q13 was located inside CIC exon 20 and that of 4q was within the coding region of the DUX4 double homeodomain gene, located within the D4Z4 repeat at the subtelomeric region of chromosome 4q (17). Reverse transcription–polymerase chain reaction (RT–PCR) analysis of tumor RNA samples was then carried out to check for chimeric transcripts containing both CIC and DUX4. As shown in Figure 3A, lanes 1 and 2, CIC–DUX4 fusions were detected in both cases; however, no reciprocal DUX4–CIC fusion was observed (Fig. 3A, lanes 3 and 4). Approximately 10 kb aberrant CIC as well as wild-type transcripts were detected in both cases by northern blotting using a CIC cDNA fragment as a probe (Fig. 3B). The aberrant signals were also detected by using a DUX4 probe (Fig. 3B). Expression of wild-type DUX4 was not detected by northern blotting or RT–PCR (Fig. 3B) (data not shown). Sequence analysis of fusion transcripts revealed that CIC was fused in frame to DUX4 and the deduced amino acid sequences of the chimeric proteins were determined (Fig. 3C). Most of the CIC protein was preserved in the chimera, including the HMG box of CIC, the putative binding site for TLE proteins, human homologs of Groucho interacting with DrosophilaCapicua (18) and highly conserved regions between human CIC and DrosophilaCapicua. Putative MAPK phosphorylation sites of CIC were also preserved. In contrast, a large part of the N-terminal DUX4 region was lost, including the DNA-binding double homeodomain. The predicted structure of the CIC–DUX4 chimera suggests that it may dysregulate its downstream target genes without altering the DNA-binding property of the CIC HMG box. Endogenous expression of the CIC–DUX4 protein was detected in the cell line ECD1 established from case 2 tumor using a polyclonal anti-CIC serum (Fig. 3D).
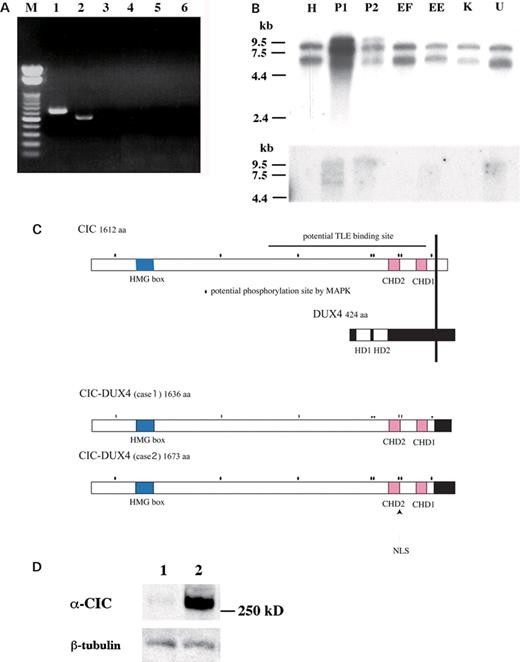
Detection of the CIC–DUX4 chimeric transcripts and the deduced structure of the chimeric proteins. (A) RT–PCR analysis of the t(4;19) tumors. CIC–DUX4 fusion transcripts were detected in two cases (lanes 1 and 2 with CIC4120 and DUX4RTr2 primers), whereas no reciprocal DUX4–CIC fusion was seen (lanes 3 and 4 with DUX4RTf1 and CICFL3 primers). Lanes 1 and 3, case 1; lanes 2 and 4, case 2 sample. M, a 100 bp ladder DNA size marker. (B) Northern blotting. Top. Aberrant large-sized signals of CIC were observed in P1 and P2 cases of t(4;19) tumors. H, HeLa cell; EF, Ewing's sarcoma with the EWS–FLI1 fusion; EE, Ewing's sarcoma with the EWS–ERG fusion; K, clear cell sarcoma with the EWS–ATF1 fusion; U, U2OS cell. Bottom. The same blot was hybridized with the DUX4 cDNA probe. Signals of same molecular size were seen in P1 and P2. Molecular sizes are indicated in kilobases. (C) Deduced structure of the CIC–DUX4 protein. CHD, Capicua homology domains; HD, homeodomain; NLS, nuclear localization signal. Putative MAPK phosphorylation sites are indicated with dots. (D) Immunoblotting for CIC–DUX4. 1, U2OS; 2, ECD1. CIC–DUX4 is detected by using an anti-CIC serum. Weak expression of the wild-type CIC is shown in U2OS cells. The anti-β-tubulin was used as control.
The CIC–DUX4 protein transforms NIH 3T3 cells and is a strong transcriptional activator
The oncogenic potential of CIC–DUX4 was determined in an NIH 3T3 cell transformation assay. The entire coding region of CIC–DUX4 was inserted into the pcDNA3.1 expression vector and introduced into NIH 3T3 cells. NIH 3T3 cells transfected with the CIC–DUX4 chimera formed significantly increased anchorage-independent colonies in soft agar compared to mock-transfected or wild-type CIC-transfected cells (Fig. 4A). The result indicates that CIC–DUX4 functions as a dominant oncogene, and the chimeric protein may act as an oncogenic transcription factor.
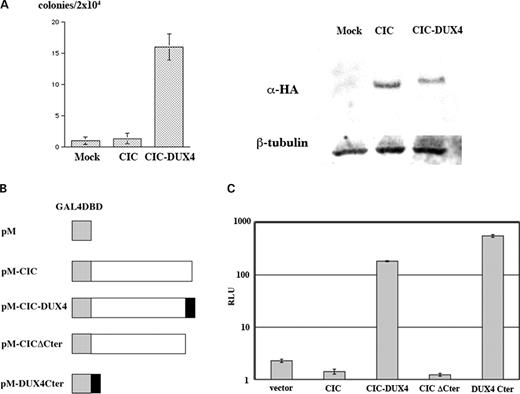
Transforming and transcriptional activities of CIC–DUX4. (A) Anchorage-independent growth of NIH 3T3 cells expressing CIC–DUX4. Numbers of colonies per 2×104 cells were determined after 2 weeks of growth in soft agar. Values are the means with standard deviation of three experiments. The right panel shows expression of HA-tagged wild-type CIC and chimeric CIC–DUX4 proteins by immunoblotting using anti-HA. (B) The pM constructs bearing the wild-type CIC, CIC–DUX4, CIC ΔC-terminus or DUX4 C-terminus. (C) The reporter assay. Enhanced luciferase activities were observed when the expression plasmids with a GAL4 DNA-binding domain fused to the full-length CIC–DUX4 or the DUX4 C-terminus were introduced. The means±standard deviations from three independent experiments are shown.
The transcriptional activity of CIC–DUX4 was then examined to clarify whether the wild-type CIC function is affected by fusion with the DUX4 sequence. cDNA fragments encoding wild-type CIC, CIC–DUX4, CIC lacking the C-terminal region or the DUX4 C-terminus were subcloned into the pM vector to obtain fusion products with the GAL4 DNA-binding domain (Fig. 4B). These constructs were co-transfected into HeLa cells with a luciferase reporter vector bearing a 5×GAL4 DNA-binding sequence. The wild-type CIC showed weak repression activity, whereas CIC–DUX4 showed 130-fold and 81-fold enhancement of transcriptional activities against the wild-type CIC and the pM vector, respectively (Fig. 4C). Because the DUX4 C-terminus showed even stronger activity (395-fold against wild-type CIC), and the repression activity of CIC ΔC-terminus was similar to that of CIC, it is very likely that the addition of the DUX4 C-terminus causes drastic modification of the CIC transcriptional activity. These results strongly suggested that the expression of CIC downstream targets might be perturbed by CIC–DUX4.
CIC–DUX4 modulates the gene expression profile
To determine whether the global gene expression profile is modulated by CIC–DUX expression, and to identify important downstream target genes, RNA samples extracted from U2OS cells with tetracycline-inducible expression of the wild-type CIC or CIC–DUX4 (Fig. 5A) were assessed by the Affymetrix GeneChip microarray system. Statistical comparisons identified 22 altered genes (30 probe sets) whose expression was significantly up-regulated in CIC–DUX4-expressing cells (Table 1). In contrast, only three genes were found to be down-regulated in CIC–DUX4 expressing cells (data not shown). This result is consistent with data obtained from the transactivation assay, and it again suggests that the chimera functions as a transcriptional activator to a majority of the target genes. Expression of 11 of the 22 up-regulated genes was validated by using quantitative RT–PCR, and significant up-regulation by CIC–DUX4 of five genes (ERM/ETV5, ETV1/ER81, MGC45780, RaLP and CCL2) both in the tet-inducible U2OS system and in the t(4;19) tumors was confirmed (Fig. 5B) (data not shown).
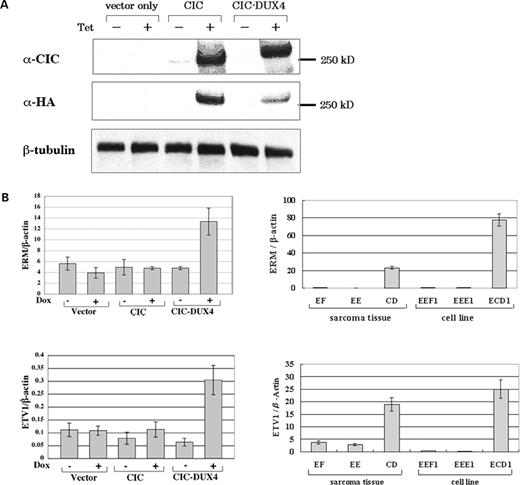
Up-regulation of PEA3 family genes by CIC–DUX4. (A) Tet-induced expression of the wild-type CIC and CIC–DUX4 proteins was detected by immunoblotting with anti-CIC serum or anti-HA. The presence of Doxycyclin is indicated by Dox+. (B) Quantitative RT–PCR. Left. Enhanced ERM (top) and ETV1 (bottom) expression achieved by tet-induced CIC–DUX4 expression is shown. Right. Up-regulation of ERM (top) and ETV1 (bottom) in the original sarcoma samples or the cell line of t(4;19) Ewing's sarcoma. EF and EEF1, Ewing's sarcoma with the EWS–FLI1 fusion; EE and EEE1, Ewing's sarcoma with the EWS–ERG fusion; CD, the t(4;19) case 1; ECD1, a cell line derived from the t(4;19) case 2. The mean expression ratios normalized against β-actin of three independent experiments are shown. Standard deviations are indicated as error bars.
Genes up-regulated by overexpression of CIC–DUX4
| Probe | Gene name | CIC–DUX4 | CIC | ||
|---|---|---|---|---|---|
| Ratio | Judge | Ratio | Judge | ||
| 229839_at | MGC45780 | 37 | Ia | 0.65 | NCb |
| 235238_at | RaLP | 9 | I | 1.3 | NC |
| 238054_at | ADPRHL1 | 5 | I | 1.3 | NC |
| 216598_s_at | CCL2 | 3.1 | I | 1.15 | NC |
| 221911_at | LOC221810 (ETV1) | 2.8 | I | 1.15 | NC |
| 226225_at | —c | 2.7 | I | 1.15 | NC |
| 224997_x_at | H19 | 2.5 | I | 2.7 | NC |
| 205630_at | CRH | 2.3 | I | 4.5 | NC |
| 206132_at | MCC | 1.9 | I | 1 | NC |
| 221011_s_at | LBH | 1.9 | I | 1.15 | NC |
| 228810_at | FLJ40432 | 1.9 | I | 1 | NC |
| 209242_at | PEG3 | 1.8 | I | 0.9 | NC |
| 203348_s_at | ERM | 1.8 | I | 1.3 | NC |
| 216375_s_at | ERM | 1.8 | I | 1.1 | NC |
| 230102_at | ERM | 1.6 | I | 1.15 | NC |
| 209392_at | ENPP2 | 1.8 | I | 1.3 | NC |
| 1555878_at | RPS24 | 1.7 | I | 0.6 | NC |
| 212012_at | D2S448 | 1.7 | I | 0.75 | NC |
| 231249_at | HT036 | 1.6 | I | 0.9 | NC |
| 227280_s_at | FLJ40432 | 1.6 | I | 0.9 | NC |
| 205048_s_at | —c | 1.6 | I | 1.15 | NC |
| 205251_at | PER2 | 1.4 | I | 1.15 | NC |
| 227433_at | KIAA2018 | 1.4 | I | 0.8 | NC |
| 225107_at | LOC442518 | 1.4 | I | 1.1 | NC |
| 228116_at | —c | 1.4 | I | 0.85 | NC |
| 205194_at | PSPH | 1.3 | I | 1.1 | NC |
| 201910_at | FARP1 | 1.3 | I | 0.85 | NC |
| 220044_x_at | CROP | 1.3 | I | 0.85 | NC |
| 36553_at | —c | 1.3 | I | 1.15 | NC |
| 225664_at | COL12A1 | 1.3 | I | 1 | NC |
| Probe | Gene name | CIC–DUX4 | CIC | ||
|---|---|---|---|---|---|
| Ratio | Judge | Ratio | Judge | ||
| 229839_at | MGC45780 | 37 | Ia | 0.65 | NCb |
| 235238_at | RaLP | 9 | I | 1.3 | NC |
| 238054_at | ADPRHL1 | 5 | I | 1.3 | NC |
| 216598_s_at | CCL2 | 3.1 | I | 1.15 | NC |
| 221911_at | LOC221810 (ETV1) | 2.8 | I | 1.15 | NC |
| 226225_at | —c | 2.7 | I | 1.15 | NC |
| 224997_x_at | H19 | 2.5 | I | 2.7 | NC |
| 205630_at | CRH | 2.3 | I | 4.5 | NC |
| 206132_at | MCC | 1.9 | I | 1 | NC |
| 221011_s_at | LBH | 1.9 | I | 1.15 | NC |
| 228810_at | FLJ40432 | 1.9 | I | 1 | NC |
| 209242_at | PEG3 | 1.8 | I | 0.9 | NC |
| 203348_s_at | ERM | 1.8 | I | 1.3 | NC |
| 216375_s_at | ERM | 1.8 | I | 1.1 | NC |
| 230102_at | ERM | 1.6 | I | 1.15 | NC |
| 209392_at | ENPP2 | 1.8 | I | 1.3 | NC |
| 1555878_at | RPS24 | 1.7 | I | 0.6 | NC |
| 212012_at | D2S448 | 1.7 | I | 0.75 | NC |
| 231249_at | HT036 | 1.6 | I | 0.9 | NC |
| 227280_s_at | FLJ40432 | 1.6 | I | 0.9 | NC |
| 205048_s_at | —c | 1.6 | I | 1.15 | NC |
| 205251_at | PER2 | 1.4 | I | 1.15 | NC |
| 227433_at | KIAA2018 | 1.4 | I | 0.8 | NC |
| 225107_at | LOC442518 | 1.4 | I | 1.1 | NC |
| 228116_at | —c | 1.4 | I | 0.85 | NC |
| 205194_at | PSPH | 1.3 | I | 1.1 | NC |
| 201910_at | FARP1 | 1.3 | I | 0.85 | NC |
| 220044_x_at | CROP | 1.3 | I | 0.85 | NC |
| 36553_at | —c | 1.3 | I | 1.15 | NC |
| 225664_at | COL12A1 | 1.3 | I | 1 | NC |
aIncrease.
bNo change.
cThe gene name has not been provided.
Genes up-regulated by overexpression of CIC–DUX4
| Probe | Gene name | CIC–DUX4 | CIC | ||
|---|---|---|---|---|---|
| Ratio | Judge | Ratio | Judge | ||
| 229839_at | MGC45780 | 37 | Ia | 0.65 | NCb |
| 235238_at | RaLP | 9 | I | 1.3 | NC |
| 238054_at | ADPRHL1 | 5 | I | 1.3 | NC |
| 216598_s_at | CCL2 | 3.1 | I | 1.15 | NC |
| 221911_at | LOC221810 (ETV1) | 2.8 | I | 1.15 | NC |
| 226225_at | —c | 2.7 | I | 1.15 | NC |
| 224997_x_at | H19 | 2.5 | I | 2.7 | NC |
| 205630_at | CRH | 2.3 | I | 4.5 | NC |
| 206132_at | MCC | 1.9 | I | 1 | NC |
| 221011_s_at | LBH | 1.9 | I | 1.15 | NC |
| 228810_at | FLJ40432 | 1.9 | I | 1 | NC |
| 209242_at | PEG3 | 1.8 | I | 0.9 | NC |
| 203348_s_at | ERM | 1.8 | I | 1.3 | NC |
| 216375_s_at | ERM | 1.8 | I | 1.1 | NC |
| 230102_at | ERM | 1.6 | I | 1.15 | NC |
| 209392_at | ENPP2 | 1.8 | I | 1.3 | NC |
| 1555878_at | RPS24 | 1.7 | I | 0.6 | NC |
| 212012_at | D2S448 | 1.7 | I | 0.75 | NC |
| 231249_at | HT036 | 1.6 | I | 0.9 | NC |
| 227280_s_at | FLJ40432 | 1.6 | I | 0.9 | NC |
| 205048_s_at | —c | 1.6 | I | 1.15 | NC |
| 205251_at | PER2 | 1.4 | I | 1.15 | NC |
| 227433_at | KIAA2018 | 1.4 | I | 0.8 | NC |
| 225107_at | LOC442518 | 1.4 | I | 1.1 | NC |
| 228116_at | —c | 1.4 | I | 0.85 | NC |
| 205194_at | PSPH | 1.3 | I | 1.1 | NC |
| 201910_at | FARP1 | 1.3 | I | 0.85 | NC |
| 220044_x_at | CROP | 1.3 | I | 0.85 | NC |
| 36553_at | —c | 1.3 | I | 1.15 | NC |
| 225664_at | COL12A1 | 1.3 | I | 1 | NC |
| Probe | Gene name | CIC–DUX4 | CIC | ||
|---|---|---|---|---|---|
| Ratio | Judge | Ratio | Judge | ||
| 229839_at | MGC45780 | 37 | Ia | 0.65 | NCb |
| 235238_at | RaLP | 9 | I | 1.3 | NC |
| 238054_at | ADPRHL1 | 5 | I | 1.3 | NC |
| 216598_s_at | CCL2 | 3.1 | I | 1.15 | NC |
| 221911_at | LOC221810 (ETV1) | 2.8 | I | 1.15 | NC |
| 226225_at | —c | 2.7 | I | 1.15 | NC |
| 224997_x_at | H19 | 2.5 | I | 2.7 | NC |
| 205630_at | CRH | 2.3 | I | 4.5 | NC |
| 206132_at | MCC | 1.9 | I | 1 | NC |
| 221011_s_at | LBH | 1.9 | I | 1.15 | NC |
| 228810_at | FLJ40432 | 1.9 | I | 1 | NC |
| 209242_at | PEG3 | 1.8 | I | 0.9 | NC |
| 203348_s_at | ERM | 1.8 | I | 1.3 | NC |
| 216375_s_at | ERM | 1.8 | I | 1.1 | NC |
| 230102_at | ERM | 1.6 | I | 1.15 | NC |
| 209392_at | ENPP2 | 1.8 | I | 1.3 | NC |
| 1555878_at | RPS24 | 1.7 | I | 0.6 | NC |
| 212012_at | D2S448 | 1.7 | I | 0.75 | NC |
| 231249_at | HT036 | 1.6 | I | 0.9 | NC |
| 227280_s_at | FLJ40432 | 1.6 | I | 0.9 | NC |
| 205048_s_at | —c | 1.6 | I | 1.15 | NC |
| 205251_at | PER2 | 1.4 | I | 1.15 | NC |
| 227433_at | KIAA2018 | 1.4 | I | 0.8 | NC |
| 225107_at | LOC442518 | 1.4 | I | 1.1 | NC |
| 228116_at | —c | 1.4 | I | 0.85 | NC |
| 205194_at | PSPH | 1.3 | I | 1.1 | NC |
| 201910_at | FARP1 | 1.3 | I | 0.85 | NC |
| 220044_x_at | CROP | 1.3 | I | 0.85 | NC |
| 36553_at | —c | 1.3 | I | 1.15 | NC |
| 225664_at | COL12A1 | 1.3 | I | 1 | NC |
aIncrease.
bNo change.
cThe gene name has not been provided.
Expression of ERM and ETV1, members of the PEA3 subclass of the ETS transcription factor gene family, was further analyzed using RNA samples derived from the original tumor tissues and cell lines. Enhanced expression of ERM and ETV1 was observed in the tumor tissue of case 1, and the cell line ECD1 established from case 2 tumor was compared with Ewing's sarcoma with respect to EWS–FLI1 or EWS–ERG fusions (Fig. 5B). Both exogenous and endogenous expressions of CIC–DUX4 were found to up-regulate ERM and ETV1. Furthermore, expression of E1AF, another member of the PEA3 family, was significantly increased in case 1 tumor tissue and in ECD1 cells (data not shown).
ERM promoter contains a CIC responsive element and is up-regulated by CIC–DUX4
To test whether ERM expression is directly regulated by CIC–DUX4, the potential promoter region of the human ERM gene was examined. A search for the genomic region 4 kb upstream of the ERM exon 1a using PROSCAN (http://bimas.dcrt.nih.gov/molbio/proscan/) revealed that there is a potential TATA box (tataaa) 309 bp upstream of the exon 1a 5′ end.
A 988 bp fragment including the potential TATA box (Fig. 6A) was subcloned into the pGL3 basic vector and the luciferase activity was measured in HeLa or ECD1 cells. Strong promoter activity was present within the fragment in ECD1 cells, whereas no activity was shown in HeLa cells (Fig. 6B, 235-fold activity in ECD1 compared with HeLa). When CIC–DUX4 was co-transfected with the reporter plasmid in HeLa cells, the luciferase activity was significantly enhanced (Fig. 6C). The enhanced luciferase activity was not observed for wild-type CIC. The reporter assay using deletion constructs showed that the 290 bp fragment indeed contains a CIC–DUX4 responsive element (Fig. 6D). Chromatin immunoprecipitation (ChIP) using the ECD1 cell line and the CIC antiserum further exhibited CIC binding on a 290 bp region inside of the CIC–DUX4 responsive element (Fig. 6E). In contrast, no CIC binding was observed at the downstream region of the responsive element. These data suggest that the region of the ERM promoter may contain a CIC recognition site that is directly responsive for and is activated by CIC–DUX4.
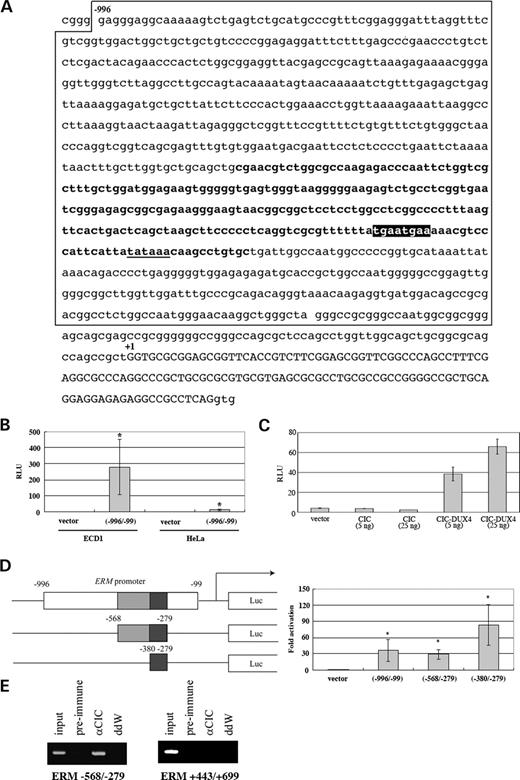
Activation of the ERM promoter by CIC–DUX4. (A) The sequence of the ERM 5′ region. The promoter region analyzed is boxed and the minimum CIC–DUX4 responsive element is indicated in bold. The consensus octameric sequence (5′-TGAATGAA-3′) for CIC HMG box is indicated as a black box. A potential TATA box is underlined. The ERM non-coding exon 1a is capitalized. (B) Strong ERM promoter activity was observed in ECD1 cells. Luciferase activities are shown as relative light intensities. An empty pGL3 promoter vector (vector) was used as a control. The means±standard deviations from three independent experiments are shown. *P<0.05. (C) The ERM promoter is activated by CIC–DUX4. The same plasmid as shown in (B) was co-transfected with indicated amounts of CIC or CIC–DUX4 expression plasmids into HeLa cells. The means±standard deviations from three independent experiments are shown. (D) The −380 to −279 region of the ERM promoter serves as a minimum CIC–DUX4 responsive element. The relative luciferase activities induced by pGL3 plasmids bearing the ERM fragments (left) are indicated as relative light units by comparing the results with the cells transfected with an empty promoter vector. The means±standard deviations from three independent experiments are shown. The nucleotide numbers from the ERM exon 1 are indicated. *P<0.05. (E) In vivo CIC binding to the ERM promoter. ChIP experiments using ECD1 cell extracts and CIC antiserum. The CIC-bound DNA fragments were immunoprecipitated with the anti-CIC or pre-immune serum and subjected to genomic PCR. The genomic PCR for the DNA sample before immunoprecipitation is shown as input. The −568/−279 promoter and +443/+699 regions of the ERM gene were amplified.
CIC–DUX4 recognizes a consensus octameric sequence and directly activates PEA3 family genes
To further evaluate direct regulation of the ERM promoter by CIC–DUX4, an electrophoretic mobility shift assay (EMSA) was carried out using a fusion protein between the CIC HMG box and glutathione S-transferase (GST), as the HMG box is a potential DNA binding-domain (19). The CIC HMG box showed specific binding to the DNA fragment within the minimal ERM promoter and the HMG box-bound DNA complex disappeared in the presence of anti-GST (Fig. 7A). The binding specificity was also confirmed by adding a cold competitor (Fig. 7B), and a CIC-binding sequence (CBS) 5′-TGAATGAA-3′ was identified. The pGL3 promoter plasmid containing triple tandem repeats of CBS was generated, and transcriptional activation by CIC–DUX4 was evident (Fig. 7C).
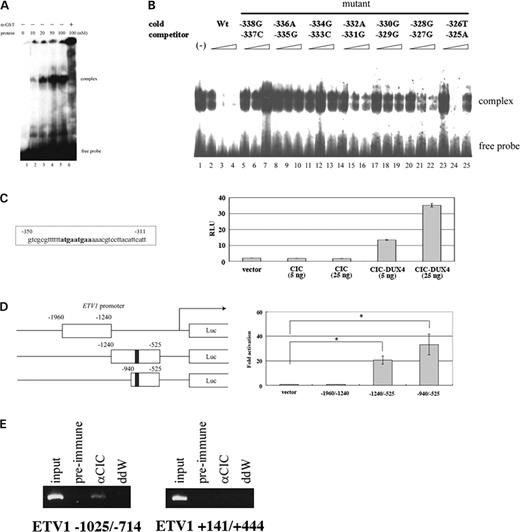
The CIC HMG box binds to the ERM promoter in vitro. (A) The DNA binding of the CIC HMG box to the −350 to −311 region of the ERM promoter. The 32P-labelled oligonucleotide was incubated with the indicated concentrations of the GST–HMG box fusion protein with or without an anti-GST antibody. The complexes were analyzed on a 5% polyacrylamide gel. (B) The DNA-binding specificity of the CIC HMG box was confirmed by using cold competitors. A 30mer probe containing the core 20 nt sequence, 5′-TATGAATGAAAAACGTCCT-3′, was incubated with 20 nm of the GST–HMG box fusion protein in the presence of the indicated amounts of the wild-type or mutant cold competitors as indicated. (C) CIC–DUX4 binds to the octameric sequence TGAATGAA. Only CIC–DUX4 enhances the promoter activity downstream to the 3×tandem repeats of the consensus octameric motif. The means±standard deviations from three independent experiments are shown. (D) CIC–DUX4 activates the ETV1 promoter. Left. The pGL3 promoter plasmids bearing the indicated ETV1 fragments were co-transfected into HeLa cells with the CIC–DUX4 plasmid. The position of the octameric sequence with 1 bp difference (TGAATgGA) was located at −921 upstream of the exon 1 (black box). Right. The −1240 to −525 and the −940 to −525 regions contained a CIC–DUX4 responsive element. CIC–DUX4 but not wild-type CIC activated the reporter. The nucleotide numbers from the ETV1 exon 1 are indicated. The means±standard deviations from three independent experiments are shown. *P<0.05. (E) In vivo CIC binding to the ETV1 promoter. ChIP experiments were performed as described in Figure 6E. The −1025/−714 promoter region of the ETV1 gene that contains 5′-TGAATgGA-3′ sequence as well as the +141/+444 regions without CBS was amplified.
The promoter region of ETV1 that is also up-regulated by CIC–DUX4 was then analyzed. Sequence analysis revealed that a 5′-TGAATGGA-3′ sequence, very similar to CBS with only a difference at the seventh nucleotide, is present 921 bp upstream of exon 1 of ETV1. Co-transfection of the reporter containing this region and the CIC–DUX4 plasmid indicated that CIC–DUX4 recognized the region as a specific promoter of ETV1 (Fig. 7D). A ChIP assay again confirmed CIC binding on a 312 bp region containing the CBS-like sequence (Fig. 7E). Furthermore, sequence analysis of the E1AF promoter region revealed that there are two octamer sequences identical to CBS 740 bp upstream of the exon 1. These data indicate that all three members of the PEA3 family and their activation regions are major targets for CIC–DUX4 by a t(4;19) translocation.
DISCUSSION
We report the identification of a CIC–DUX4 fusion that was found in two cases of Ewing-like sarcoma bearing the recurrent t(4;19)(q35;q13). The t(4;19) is a rare chromosomal translocation, not appeared as a recurrent translocation in a public database (http://cgap.nci.nih.gov/Chromosomes/Mitelman). No similar chromosomal abnormality has been observed in 600 cases of malignant bone and soft tissue tumors examined in our institute. However, a case of primitive mesenchymal tumor with t(4;19)(q35;q13.1) developed in a 12-year-old boy was reported (20). The similar translocation was also detected in rhabdomyosarcoma with complex chromosomal aberrations (21,22). Thus, t(4;19) translocation may be a genetic hallmark of a subclass in small round cell tumors, despite its rare occurrence.
The C-terminal region of DUX4 becomes fused to CIC, and CIC–DUX4 acquires a transforming activity against NIH 3T3 fibroblasts, indicating that CIC–DUX4 acts as a dominant oncogene. The fusion of the DUX4 sequence to CIC provides a strong transcriptional activity. There was no significant difference of intracellular localization between the wild-type CIC and CIC–DUX4 (data not shown). Therefore, it is intriguing to clarify the protein components of CIC- and CIC–DUX-DNA binding complexes.
The DUX4 fusion to CIC up-regulates PEA3 family genes, which might play an important role in tumorigenesis. Although PEA3 family genes were identified using expression profiling in U2OS osteosarcoma cells, high-level expression of PEA3 genes has been confirmed in the original tumor samples. Two of three PEA3 family genes, ETV1 and E1AF, are fused to EWS in EFTs with t(7;22) and t(17;22), respectively (7,8), and it is considered that functions of ETS proteins, including PEA3 proteins, are enhanced upon fusion with EWS (15). Up-regulation of PEA3 family genes by CIC–DUX4 is therefore an equivalent molecular change to the EWS–ETS fusion, and Ewing's sarcoma with the CIC–DUX4 fusion does not require an EWS–ETS fusion.
There were close similarities including shared pathological features between the present cases and EFTs with conventional EWS–ETS fusions. These patients were somewhat older than the characteristic age range seen for EFTs, and CD99/MIC2 expression in these cases was weaker than that typical of Ewing's tumor cells. These tumors might belong to a novel disease category; however, up-regulation of PEA3 family genes by CIC–DUX4 indicates a close similarity in molecular pathogenesis between t(4;19) tumors and EFTs.
Up-regulation of PEA3 family genes has been reported in a series of human malignant neoplasms such as breast, ovarian and gastrointestinal cancers, and their overexpression is related to invasive and metastatic phenotypes (22–26). More recently, ETV1 has been found fused to androgen-driven TMPRSS2 in human prostatic cancers (27). Together with these data, our present study underscores the importance of the PEA3 family in tumorigenesis and tumor progression in a broad spectrum of human malignancies.
CIC is a human homolog of the Drosophila capicua gene product that has been identified as a transcriptional repressor of the torso receptor tyrosine kinase (RTK) pathway (18). The repression activity of capicua against tailless and huckebein is achieved by interaction with the co-repressor groucho, and the RTK signal dissociates the interaction between capicua and groucho resulting in precise posterior patterning (18,28). In addition, capicua acts as a positive regulator for pipe in follicle cells (29). Although the physiological function of this protein in human CIC remains to be clarified, it is expressed in developing cerebellar neurons and is involved in ErbB signaling (30,31). Our present study has indicated that CIC functions as a sequence-specific transcription factor by binding TGAATG(G/A)A, although the binding capability is modest, as in the case of other HMG box proteins (32,33). The homology of the HMG box with CIC and other HMG family proteins is not very high, and the identified consensus sequence is different from the canonical CC(T/A)TTG(T/A)(T/A)CT of LEF1, SRY or ROX1 (19), suggesting that CIC may belong to a novel subfamily of HMG box proteins.
DUX4 is located within the D4Z4 sequence, which is a 3.3 kb tandem repeat located at the subtelomeric region of 4q as well as of other chromosomes including 10q, 21q and 22q (34). The translocation breakpoint at 4q35 is closely linked to facioscapulohumeral muscular dystrophy (FSHD), and the copy number of D4Z4 is significantly reduced in FSHD patients (17). The biological function of DUX4, however, remains unclear, and its expression has not been clearly demonstrated yet (35). Our report is the first indication that the gene at the FSHD locus is in fact involved in human diseases. It has been proposed that D4Z4 may function as a cis-acting element and that the reduced number of D4Z4 may affect expression of other nearby genes (36). This hypothesis is fascinating because the 4q35 with the normal number of the D4Z4 repeats may repress the genes located around the 19q13 break such as ERF, which encodes the ETS repression factor. However, our preliminary experiments failed to show significant changes in gene expression of ERF, GSK3A, PAFAGH1B3 or EGFL4 (Fig. 2C) between t(4;19) tumors and conventional Ewing's sarcomas (Nakamura, T., unpublished data). Alternatively, a more dynamic structural alteration such as chromosomal looping (37) may affect expression of 19q genes located at more distant regions. Together with the earlier investigations, our present study indicates that the major oncogenic effect of the t(4;19) translocation is alteration of the transactivating potency of the CIC–DUX4 chimera and the DUX4 C-terminus contributes to this increased potency.
MATERIALS AND METHODS
Patients
Case 1
An invasive tumor originated in the muscle tissue of the left buttock of a 62-year-old female patient, forming a large tumor mass in the pelvic space with vaginal invasion. A representative histological image is shown in Figure 1. The tumor shown is diffuse proliferation of undifferentiated small round tumor cells (Fig. 1A). The tumor cells had round nuclei and clear and scant cytoplasms and contained a small number of PAS-positive glycogen granules (Fig. 1B). No rosette was seen. Immunohistological examination revealed that the tumor cells were weakly positive for CD99/MIC2 and positive for vimentin (Fig. 1C) (data not shown). A curative surgical operation was not carried out, and irradiation and chemotherapy had little effect. The patient died of the disease 10 months after the biopsy. The tumor karyotype was characterized as a reciprocal translocation t(4;19)(q35;q13) in 10 out of 10 metaphase tumor cells examined [53–55, XX, +2, t(4;19)(q35;q13), +6, +8, +13, +16, +17, del(19)(q13), +20].
Case 2
A soft part tumor, 80 mm in diameter, developed in the left shoulder of a 31-year-old male patient. After chemotherapy with cisplatin and adriamycin and irradiation, the tumor was completely resected. The patient has been healthy without recurrence for 30 months after the operation. Histologically, the tumor was composed of a densely packed growth of small round cells with round nuclei and scant cytoplasms with glycogen granules. The tumor cells were positive for vimentin and were weakly positive for CD99/MIC2. As in case 1, a reciprocal t(4;19)(q35;q13) translocation was seen in 10 out of 10 metaphase tumor cells examined [48, XY, t(4;19)(q35;q13), +6, +16].
Fluorescence in situ hybridization
Nine BAC clones located within the 4.7 Mb region of 19q13 (Fig. 2A) were purchased from Research Genetics and used for FISH analysis. All BACs were labeled directly with SpectrumGreen-dUTPs by nick translation according to manufacturer's recommendation (Vysis, Dowers Grove, IL, USA). The CEP4 probe that recognizes the centromeric region of chromosome 4 was purchased from Vysis. The probes were blocked with Cot-1 DNA (Vysis) to suppress repetitive sequences. Metaphase spreads obtained from patient's cell line were hybridized at 37°C overnight with labeled probes. After post-hybridization washes, the chromosomes were counterstained with DAPI. Cell images were captured using Axiophoto-2 fluorescent microscopy (Carl Zeiss, Jena, Germany) and a CCD camera (Photometrics SenSys camera) connected to a computer running the Quips Imagecapture analysis system (Signal Analytics, Vienna, VA, USA).
DNA extraction and Southern blot analysis
DNA extraction, restriction endonuclease digestion, agarose gel electrophoresis, Southern blot transfer and hybridization were carried out as described previously (38). A 710 bp DNA fragment within the CIC exon 10 was used as a probe to detect rearrangements (Fig. 2C).
Cloning of the chimeric gene
A subgenomic DNA library was constructed using tumor DNA and the ZAP Express phage vector (Stratagene, La Jolla, CA, USA). The library was screened with a CIC DNA probe according to the method described previously (39). A single clone containing an inserted 5.5 kb DNA fragment corresponding to the BamHI rearranged fragment was isolated and sequenced using the Dye Terminator Cyclesequencing Kit (Beckman Coulter, Fullerton, CA, USA) and a CEQ8000 DNA sequencer (Beckman Coulter). A BLAST search was carried out using the public database available at the University of California at Santa Cruz (http://genome.ucsc.edu).
Reverse transcription–polymerase chain reaction
RT–PCR was carried out as previously described (40). The PCR primers for CIC and DUX4 were as follows. CIC4120: 5′-TGAGTTGCCTGAGTTTCG-3′, CICFL3: 5′-AGGGGTCCCTCACCTGCCTGTGGCAGCTG-3′, DUX4RTr2: 5′-TGAGGGGTGCTTCCAGCG-3′, DUX4 RTf1: 5′-ACAGGGGGCTTTCGTGAG-3′.
Northern blotting
Poly(A)+ RNA isolation, gel fractionation, membrane transfer and hybridization were carried out as previously described (40). A 700 bp cDNA fragment derived from CIC exon 10 was used as a probe.
Cell culture
The ECD1 cell line was established from tumor tissue of case 2. The EEF1 and EEE1 cell lines were established from Ewing's sarcoma with EWS–FLI1 or EWS–ERG chimeras, respectively. These cell lines were grown in RPMI-1640 medium (Sigma, St Louis, MO, USA) supplemented with 50 U/ml penicillin, 0.1 mg/ml streptomycin and 10% fetal bovine serum. NIH 3T3 and U2OS cells were grown in Dulbecco's modified Eagle's medium (Sigma) supplemented with 50 U/ml penicillin, 0.1 mg/ml streptomycin and 10% fetal bovine serum.
Transformation assays
Wild-type CIC and CIC–DUX4 cDNAs were isolated from the ECD1 cell by RT–PCR. The PCR products were subcloned into plasmids and the sequences were verified by analyzing multiple independent clones. No mutation other than the fusion was observed. The full coding regions of CIC and CIC–DUX4 were then HA-tagged and subcloned into the pcDNA3.1 vector (Invitrogen). Transfection of plasmids and transformation assays were performed as described previously (40).
Inducible expression of CIC–DUX4 in U2OS cells
The HA-tagged CIC and CIC–DUX4 were subcloned into the pcDNA4/TO/myc-His vector (Invitrogen). TRExU2OS cells (Invitrogen) were transiently transfected with plasmid DNAs using Lipofectamine (Invitrogen), and 24 h after transfection, cells were treated with 1 mg/ml of Doxycyclin (Sigma) to obtain expression of CIC or CIC–DUX4. Cells were grown for a further 24 h and total RNA was isolated by using the RNAeasy mini kit (QIAGEN, Hilden, Germany).
Western blot analysis and antibody
Western blot was performed as previously described (40). A rabbit polyclonal anti-CIC antibody was raised against a peptide consisting of CIC amino acids 1362–1567. Anti-ERM (H-100) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Array hybridization
The oligonucleotide array Human Genome U133 Plus 2.0 (Affymetrix, Santa Clara, CA, USA), composed of 38 500 human genes and ESTs, was hybridized with cRNA probes generated from TRExU2OS cells according to manufacturer's instructions. Arrays were scanned using an Affymetrix confocal scanner and analyzed using Affymetrix MicroArray Suite version 5.0 software (Affymetrix). Intensity values were scaled so that the overall fluorescence intensity of each chip of the same type was equivalent. A gene whose expression was over 1.5-fold higher or lower in CIC–DUX4 expressing cells than control cells was judged to be a gene with modified expression.
Quantification of ERM and ETV1 expression by real-time RT–PCR
To quantify expression of ERM and ETV1, SYBR Green real-time PCR (QIAGEN) and a 7900HT sequence detector (Applied Biosystems, Foster City, CA, USA) were used. Reactions contained 2×QuantiTect SYBR Green PCR Master Mix and 200 nm of forward and reverse primers (ERM forward: 5′-AGGGAAATCTCGATCTGAGGAATG-3′, ERM reverse: 5′-GCTAACCAAGCCTCTTGAAGTTGAC-3′; ETV1 forward: 5′-TGGGGCATTCAGAAAAACAGG-3′, ETV1 reverse: 5′-TGTCCTCCTCGTTGATGTGACG-3′). β-actin expression served as an internal control. Thermal cycling proceeded with 45 cycles of 95°C for 15 s, 55°C for 30 s and 72°C for 30 s. The amounts of input RNA were calculated using relative standard curves for the mRNAs of ERM, ETV1 and β-actin.
Luciferase assay
A 908 bp fragment of the ERM promoter and a 1.4 kb fragment of ETV1 were amplified by PCR using human genomic DNA and were subcloned into the pGL3-Basic or pGL3-Promoter vectors (Promega). The wild-type CIC, CIC–DUX4 chimera, CICΔC and DUX4 C-terminus were subcloned into the pM vector to obtain fusion proteins with the GAL4 DNA-binding domain, as previously described (41). A total of 4×105 HeLa cells were co-transfected with 0.5 µg of the reporter plasmid, 2 ng of pRL-TK and 0.1 µg of expression plasmids unless otherwise specified in the figure legends. Luciferase activities were measured using the dual luciferase reporter assay system (Promega) and Lumat LB 9507 (Perkin Elmer).
Chromatin immunoprecipitation
ChIP for the ERM promoter region was performed as described (42). Briefly, ECD1 cells (2×108) were cross-linked in 20 ml of paraformaldehyde buffer (0.2% paraformaldehyde, 10 mm PBS, 150 mm NaCl, 0.25% Triton X-100) for 10 min. After washing, cells were sonicated three times for 30 s each at the maximum settings (U 200S, IKA LABORTECHNIK, Staufen, Germany) followed by centrifugation for 15 min at 10 000g. Immunoprecipitation was performed with specific antibody (CIC 1341 A.P) and salmon sperm DNA. Immunoprecipitated DNA fragments were purified with a QIAquick Spin Kit (QIAGEN), and PCR was carried out using the primers ERM g-568MluIF; 5′-GTAACGCGTATAACTTTGCTTGGTGCTGCAGCTGCG-3′ ERM g-279BglIIR; 5′-CATAGATCTCCATTGGCCAATCAGCACAGGCTTG-3′.
Electrophoretic mobility shift assay
A GST fusion protein was made by subcloning the cDNA sequence corresponding to the CIC HMG box into the pGEX6P vector (Amersham). A 100 bp DNA fragment of the ERM promoter or a 30 bp oligonucleotide probe (5′-gtttTTATGAATGAAAAACGTCCTtacat-3′) was labeled with [α-32P]dCTP by fill-in reaction or [γ-32P]ATP by end-labeling, respectively. EMSA was performed as described previously (43). Probes were incubated with purified HMG box or control GST proteins with or without anti-GST antibody (Amersham).
ACKNOWLEDGEMENTS
The authors thank Takeshi Kuwata, Ichizo Nishino and Tsuyoshi Ishida for useful comments. This work was supported by a Grant-in-Aid for Scientific Research for Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Conflict of Interest statement. None declared.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}