
Abstract
Among the recent advances in the molecular targeted therapy of cancer, the applications focused on epidermal growth factor receptor (EGFR) are currently the most promising and the most advanced at clinical level. In view of the different modes of action of monoclonal antibodies and tyrosine kinase inhibitors (TKI), it is tempting to examine the effect of a combination between these two EGFR targeting approaches. It was the purpose of the present study to test this combination at experimental level by using two epidermoid human cell lines CAL 33 and CAL 39. As C225 (Cetuximab®) and ZD1839 (Iressa®) are, respectively, the most clinically advanced drugs in the category of anti-EGFR drugs, the experiments were performed using these two representative compounds. The combination of C225 and ZD1839 was antagonistic whatever the cell line considered. These antagonistic effects were corroborated by molecular changes in apoptosis (PARP) and EGFR signalling (phospho-p42–44). Drugs alone led to a diminution in EGFR levels, while their combination increased the cellular expression in EGFR. These data suggest that new and tempting treatment strategies on the EGFR target consisting in a double hit with a monoclonal antibody and a TKI must be considered with caution.
Similar content being viewed by others

Main
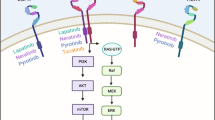
Epidermal growth factor receptor (EGFR) is a protein tyrosine kinase which plays a crucial role in signal transduction pathways that regulate key cellular functions such as survival and proliferation. Among the recent advances in the molecular targeted therapy of cancer, the applications focused on EGFR are currently the most promising and the most advanced at clinical level (Arteaga, 2001; Baselga, 2001; Ciardiello and Tortora, 2001; Yarden, 2001; Mendelsohn, 2002; Zwick et al, 2002). Considering the set of therapeutic tools targeting EGFR (Modi and Seidman, 2002), there are at present two well-identified emerging categories of drugs, the one characterised by monoclonal antibodies (Mabs) and the other by tyrosine kinase inhibitors (TKIs).
Mabs and TKIs clearly differ in their mode of action at target level. The primary action mechanism of C225, a chimeric Mab, is a competitive antagonism for EGFR. Independent of the phosphorylation status of the receptor, the EGFR–C225 complex is subsequently internalised (Fan et al, 1994; Prewett et al, 1996). The outcome of the EGFR–C225 complex following internalisation is not clearly documented, particularly regarding the stage between degradation and cell membrane recycling of the intact receptor. Tyrosine kinase inhibitors act on the cytosolic ATP-binding domain of EGFR by inhibiting EGFR autophosphorylation. Depending on the nature of the TKI, EGFR inhibition can be either reversible, as with ZD839 or OSI-774, or irreversible, as for instance with PD183805 (Noonberg and Benz, 2000; Ciardiello and Tortora, 2001; Denny, 2002). The irreversibility of the inhibition is due to covalent fixation of the drug at the ATP-binding site. In contrast to the approach using Mabs, the use of TKIs is not strictly specific for the ATP pocket of the EGFR; this can be explained by the fact that TKIs are all ATP competitors at the ATP site of the tyrosine kinases (Denny, 2002). Thus, for TKIs, some variable crossreactivity may exist between EGFR and other HER-B family members such as HER-2 (Arteaga, 2001).
In view of the different modes of action of Mabs and TKIs on EGFR, it is tempting to examine the effect of a combination between these two EGFR-targeting approaches. In addition, clinical studies indicate that the pharmacodynamics of Mabs and TKIs are not strictly superimposable with side effects as regards Mabs being mainly centred on the skin and, regarding TKIs, mostly related to the skin and to gastrointestinal intolerance (Thomas and Grandis, 2004). It was the purpose of the present study to test this combination at the experimental level by using two epidermoid human cell lines CAL 33 and CAL 39. As C225 (Cetuximab®; Herbst and Hong, 2002; Saltz et al, 2004) and ZD1839 (Iressa®; Ranson et al, 2002; Fukuoka et al, 2003) are, respectively, the most clinically advanced drugs in the category of Mabs and TKIs, the experiments were performed using these two representative compounds. Along with an analysis of the drug impact on cell proliferation, the present study included an examination of drug effects on apoptotic pathway (phospho-AKT and PARP cleavage), on MAP kinase pathway (phospho-MAPK) and on EGFR expression.
Materials and methods
Chemicals
All chemicals were obtained from Sigma Chemicals (Saint Quentin Fallavier, France) and were of the highest purity grade. DMEM and glutamine were obtained from Biowhittaker (Verviers, Belgium) and foetal bovine serum (FBS) from Dutscher (Brumath, France). Drugs were kindly provided: Cetuximab® (C225) by Merck (Darmstadt, Germany) and Iressa® (ZD1839) by Astra Zeneca (Rueil Malmaison, France), respectively.
For ZD1839, a 50 mM working solution in dimethylsulphoxide (DMSO) was prepared before use. Radiochemicals, 3H-thymidine and 125I-EGF, were provided by Amersham Biosciences (Orsay, France).
Cell lines
One human head and neck cancer cell line CAL33 and one human vulvar cancer cell line CAL39 were used in the present study. Epidermal growth factor receptor levels (fmol mg−1 prot−1) were 33 800 and 6500 for CAL33 and CAL39, respectively (ligand-binding assay as previously published by us; Olivier et al, 1990). Both cell lines were maintained in DMEM supplemented with 10% FBS, 2 mM glutamine, 50 000 U l−1 penicillin and 80 μ M streptomycin in a fully humidified incubator (Sanyo, Japan) at 37°C in an atmosphere containing 8% CO2.
Drug administration schedule
Drug effects were assessed using 3H-thymidine incorporation. We checked whether this assay compared well with the viability results obtained with the classical MTT test: the results were almost superimposable for ZD1839; it was not the case for C225, which exhibited acceptable dose–effect curves with the thymidine incorporation only. Experiments were performed in DMEM supplemented with 0.5% FBS. This low serum concentration was selected so as to reduce the presence of exogenous growth factors while ensuring a correct survival of cells without drug. Cells were seeded on day 0 in a 24-well microplate, 18 000 cells per well for CAL33 and 24 000 cells per well for CAL39. The cell seeding concentrations were chosen to allow exponential growth throughout the experiment. Drugs were added 48 h after cell seeding and applied during 96 h either alone or in combination. The range of drug concentrations was chosen according to the results of preliminary experiments in order to obtain the most complete single-agent dose–effect curves. Concentrations thus varied between 10−11 and 10−7 M for C225 and between 10−8 and 10−4 M for ZD1839. The assay method was essentially that described by Volm et al. (1970): after the end of the drug incubation time, that is, 120 h after cell seeding, [3H]dTHd was added (52 CimM−1; final concentration, 3 μCi ml−1) and incubation was continued for another 2 h. Plates were then cooled on ice. The experiments were ended by adding ice-cold 5% trichloracetic acid (TCA) and the wells were washed twice with TCA, then precipitated DNA was dissolved in NaOH 10% solution and counted on a liquid scintillation counter (Wallac, 1409 DSA). Results were expressed as the percentage of radioactive incorporation relative to a control without the drugs. Experiments were performed at distance in triplicate.
Drug interaction analysis
Combination Index (CI) calculations
The cytotoxic effects obtained with the ZD1839/C225 combinations were analysed according to the Chou and Talalay method (1984) on Calcusyn software (Biosoft, Cambridge, UK). Interaction between ZD1839 and C225 was assessed by means of an automatically computed CI.
The use of the Chou and Talalay ‘CI’ requires a dose–effect curve, which reaches the zero value for survival. This was not strictly the case for the C225 dose–effect curve. For this reason, in a complementary analysis, we replaced the CI by the following ratio R:

for example, if C225 at a concentration [c] and ZD1839 at a concentration [z] which, when given alone, have a 50% growth-inhibitory effect and, when given together (at these same concentrations, respectively, [c] and [z]), have a 75% effect, then in this case R will be: (1–0.75)/0.5 × 0.5=1.
Then, if R<0.8, the association is considered to be synergistic

Molecular factors
All experiments concerning the following investigated molecular factors were performed on CAL33 cells.
Epidermal growth factor receptor measurement by ligand-binding assay
Epidermal growth factor receptor expression was assayed as previously described by us (Olivier et al, 1990). Cells were grown to 80–90% confluence, in 24-well plates, in 10% FBS–DMEM at 37°C. Then drugs (ZD1839 2 × 10−6 M, C225 10−9 M final concentration) were added and incubation was performed during 2 days. Cells were rinsed three times with 500 μl RPMI 1640 containing 0.1% BSA at 2–4°C and incubated for 30 min with the same medium (500 μl per well) at 4°C. Epidermal growth factor receptor content was determined by incubation in RPMI medium for 3 h at 4°C in the presence of increasing concentrations of 125I-EGF (0.01, 0.02, 0.04, 0.08, 0.12, 0.18, 0.2 nM) and with 0.2 nM of 125I-EGF with increasing concentrations of unlabelled EGF (0.05, 0.1, 0.2, 0.4, 0.8, 1.6, 3.2, 6.4, 20, 200 nM). Plates were put on ice to stop the reaction, the supernatant was removed, and cells were washed twice with PBS containing 0.1% BSA (4°C, 500 μl per well). Cells were solubilised in 1 M NaOH at 37°C (500 μl per well for 30 min). Radioactivity was determined by gamma counting. The number of receptor sites per cell (N) and the dissociation constant (Kd) were determined by Scatchard analysis (each point on the Scatchard plots was plotted in quadruplicate). Cell number was determined in wells run in parallel, by resuspending cells in 200 μl PBS at room temperature and counting with a haemocytometer. Experiments were performed at distance in duplicate.
Western blot analyses
Cells were seeded in a 75 cm2 cell culture flask for 48 h in DMEM+10% FBS medium. They were then washed three times with PBS and incubated for another 48 h in DMEM medium without FBS in the presence of 2 × 10−6 M ZD1839 and/or 1 × 10−9 M C225 (concentrations at which drug maximum effect on thymidine incorporation was obtained). Then, at the end of drug exposure, serum (FBS, 10% final concentration) was added and cell pellets were collected at t0 (before addition of serum) and 5 and 30 min after addition of FBS. All cell pellets were stored at −80°C before analysis. Cell pellets collected at t0 were used for the measurement of cleaved PARP and EGFR; those obtained at 5 and 30 min were intended for the analysis of PAKT and Pp42–44. The expression of EGFR, HER-2, cleaved PARP, PAKT, Pp42–44 was measured using the Western blot technique. Briefly, equivalent quantities of protein for each drug combination were separated on a 12% SDS–polyacrylamide gel (EGFR and HER-2 excepted, for which a 7.5% polyacrylamide gel was used). After overnight transfer to a membrane, the proteins were revealed: for PAKT, the first antibody was rabbit polyclonal (Ozyme) 1/1000 in TBS 5% BSA using Jurkat cells as a positive control. For Pp42–44, the first antibody was of mouse origin kindly provided by CNRS Unit 6543 (Centre Antoine Lacassagne, Nice) 1/5000 in TBS 5% milk using A431 as a positive control. For PARP, the first antibody was of Rabbit origin (Ozyme) 1/1000 in TBS 5% milk. For EGFR, the first antibody was the mouse monoclonal Ab12 (Neomarkers) 1/1000 in TBS 5% milk using A431 as a positive control. After blotting with an adapted second antibody, proteins were revealed in a darkroom using ECL. All experiments were performed in triplicate.
Results
The effects of C225, ZD1839 and their combinations are described in Figures 1 and 2 for CAL33 and CAL39 cells, respectively. Of note, and for both cell lines, C225 alone never led to 0% survival. The dose–effect curves of the C225+ZD1839 combination were either just under (CAL 33 cells) or even clearly above (CAL39 cells) the ZD1839 dose–effect curve. This direct observation suggests an antagonistic interaction between the two drugs. The examination of both R values and CIs confirmed that the combination of C225 and ZD1839 was antagonistic whatever the cell line considered (Table 1 ).

Dose–effect curves of C225 alone, ZD1839 alone and their combination on CAL33 cell line. Bars depict standard deviations from triplicate experiments.

Dose–effect curves of C225 alone, ZD1839 alone and their combination on CAL39 cell line. Bars depict standard deviations from triplicate experiments.
The effects of C225, ZD1839 and their combination on PARP, PAKT and Pp42–44 are depicted in Figure 3. The impact of ZD1839 on PARP cleavage was more marked than that generated by C225 alone (Figure 3A). Of note, when the two drugs were combined, there was a smaller change in PARP as compared to the change with ZD1839 alone. This finding is in agreement with the data on cell survival indicating infra-additive cytotoxic effects resulting from the association of the two anti-EGFR drugs. There were no clear-cut effects of ZD1839, C225 or their combination on PAKT (Figure 3B). ZD1839 or C225 given alone downregulate Pp42–44 up to 30′ after serum addition; in contrast, combining them clearly enhanced this cell division-related pathway (Figure 3C). This observation corroborates the results on cell proliferation and strengthens the antagonistic interaction for cell survival between ZD1839 and C225.

(A) Effects of C225, ZD1839 and their combination on PARP cleavage after the end of drug exposure and before serum input on CAL33 cell line. Bars depict standard deviations from triplicate experiments. (B) Effects of C225, ZD1839 and their combination on PAKT expression induced by serum input on CAL33 cell line. Bars depict standard deviations from triplicate experiments. (C) Effects of C225 and ZD1839 on the P-p42–44 expression induced by serum input on CAL33 cell line. Bars depict standard deviations from triplicate experiments.
The apparent antagonism between the two drugs could be accounted for by a modification of EGFR sites. Epidermal growth factor receptor cell quantification and EGFR ligand affinity are depicted in Table 2 and Figure 4 for the different tested conditions. Drugs alone led to a diminution in EGFR levels as compared to controls, with a stronger effect for C225 as compared to ZD1839. In contrast, the combination of the two drugs increased the cellular expression in EGFR. This increase was observed with both EGFR protein measurement (Figure 4) and with the specific quantification of EGFR receptors (Table 2).

Effects of C225, ZD1839 and their combination on EGFR expression on CAL33 cell line. Bars depict standard deviations from triplicate experiments.
Discussion
In the current search for new therapeutic targets in cancer, growth factors and their receptors represent a field of active investigation both at preclinical and clinical levels. The EGFR receptor is significantly overexpressed in solid tumors and constitutes an important target for the development of the new targeted anticancer drugs (Baselga, 2001; Mendelsohn, 2002). There are two different categories of compounds in the current arena of active drugs targeting EGFR with monoclonal antibodies on the one hand and TKIs on the other. C225 (cetuximab) is the most clinically advanced representative drug in the family of monoclonal antibodies targeting EGFR (Herbst and Hong, 2002; Saltz et al, 2004). ZD1839 (Iressa) is one of the EGFR-specific TKIs currently under active clinical investigation (Ranson et al, 2002; Fukuoka et al, 2003). Although C225 and ZD1839 target the same EGFR receptor, there are some differences between these two drugs. First, they interact with different domains of the EGFR protein with C225 recognising the extracellular part of EGFR and ZD1839 acting on the intracellular ATP pocket of the tyr kinase entity. As the biological activity of ZD1839 may be attributed to its antityr kinase action, the exact mechanism of action of C225 is perhaps more complex. Its activity is partly related to its interaction with the EGFR and partly related to antibody-directed cell cytotoxicity as described for Herceptin (Clynes et al, 2000). C225 is administered by i.v. injections, while ZD1839 is an oral drug. The pharmacodynamics of the two drugs, although superimposable regarding adverse cutaneous effects, differ as regards digestive toxicity, which is mainly observed during ZD1839 treatment (Thomas and Grandis, 2004). Of interest, the two drugs may possess different resistance mechanisms, since it has recently been reported that the clinical efficacy of Iressa in lung cancer patients was related to the presence of activating mutations located in the domain of the ATP pocket (Lynch et al, 2004); it is not certain that these mutations impact on the antitumour efficacy of monoclonal antibodies. Taken together, the above-mentioned arguments provided justification for testing the combination of C225 and ZD1839. A recent study by Huang et al (2004) examined the antitumour effects resulting from this dual combination more marked tumour regressions were observed with the combination of ZD1839 and C225 in mice bearing a human lung cancer xenograft. Another recent study by Matar et al (2004), based on both in vitro and in vivo data, led to similar conclusions.
In the present study, when combining data from cell survival and those obtained by examining molecular factors, there are strong concording arguments suggesting that the combination of the two drugs triggers less than additive cytotoxic effects. The studies by Huang et al (2004) and Matar et al (2004) were based on in vitro and in vivo experiments. It must be underlined that, when examining the different cell lines which were explored in these two latter studies, the supra-additivity of the dual EGFR targeting was not found in all explored cell lines; these cell lines differed markedly between them for the EGFR content. Differences in intrinsic EGFR tumoral expression may modulate the final impact of the dual EGFR targeting and explain the differences between the present conclusions and those reported by the two other groups (Huang et al, 2004; Matar et al, 2004). In the present study, the infra-additive impact on cell survival was sustained by the changes in cleaved PARP, a faithful molecular indicator of apoptotic process, showing that C225-ZD1839 caused less apoptosis than ZD1839 alone. Further drug-related specific molecular examination indicated that, following drug exposure and cell stimulation by the medium, there was very little activation of the Map kinase pathway (changes in P-p42–44) following cell treatment by either drug, contrasting with the sharp increase in P-p42–44 noted after the combined application to ZD1839 plus C225 (Figure 3c). This observation could plausibly be explained by the fact that C225, markedly, and ZD1839, slightly, downregulate EGFR expression (Figure 4), while their combination has a marked opposite effect with an overexpression of EGFR. Importantly, both analytical methods (Western blot and ligand-binding assay) concurred to highlight the upregulation of EGFR when administering the drug combination. This means that the increase in EGFR involves active and functional receptors (data from Scatchard analysis). The underlying mechanism of this upregulation of the EGFR target produced by the two drugs is not easy to elucidate. Receptor downregulation has been studied most effectively for tyrosine kinase receptor and especially for EGFR (Waterman and Yarden, 2001). Thus, subsequent to its ubiquitination, EGFR is subject to lysosomal degradation (Citri et al, 2002). It has been reported that the binding of the natural ligand to EGFR results in a conformational change in the external domain of the receptor (Greenfield et al, 1989), which could be crucial to the ligand-induced internalisation of the receptor (Opresko et al, 1995). There is thus a ligand-controlled turnover in the expression of EGFR, which could be deregulated in the combined presence of C225 and ZD1839. Recently, the identification of proteins p70 and Clip 4 was reported (Kowanetz et al, 2004); these proteins inhibit endocytosis of EGFR and interact with Cbl, a ubiquitin ligase which plays a critical role in EGFR endosomal degradation (Ettenberg et al, 2001). It is possible that the combined presence of C225 and ZD1839 may alter the interaction of Cbl with EGFR by a conformational change induced in EGFR.
In conclusion, the present study provides concording pre-clinical data based on cell toxicity and molecular pharmacology. These data suggest that new and tempting treatment strategies on the EGFR target consisting in a double hit with a monoclonal antibody and a tyr kinase inhibitor must be considered with caution.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Arteaga CL (2001) The epidermal growth factor receptor: from mutant oncogene in non-human cancers to therapeutic target in human neoplasis. J Clin Oncol 19: 324–340
Baselga J (2001) Targeting the epidermal growth factor receptor: a clinical reality. J Clin Oncol 19: 41–44
Chou T, Talalay P (1984) Quantitative analysis of dose–effects relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22: 27–55
Ciardiello F, Tortora G (2001) A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res 7: 2958–2970
Citri A, Alroy I, Lavi S, Rubin C, Xu C, Grammatikakis N, Patterson C, Neckers L, Fry DW, Yarden Y (2002) Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. EMBO Journal 21: 2407–2417
Clynes RA, Towers TL, Presta LG, Ravetch JV (2000) Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med 6: 443–446
Denny WA (2002) Irreversible inhibitors of the erbB family of protein tyrosine kinases. Pharmacol Ther 93: 253–261
Ettenberg SA, Magnifico A, Cuello M, Nau MN, Rubinstein YR, Yarden Y, Weissman AM, Lipkowitz S (2001) Cbl-b-dependent coordinated degradation of the epidermal growth factor receptor signaling complex. J Biol Chem 276: 27677–27684
Fan Z, Lu Y, Wu X, Mendelsohn J (1994) Antibody-induced epidermal growth factor receptor dimerization mediates inhibition of autocrine proliferation of A431 squamous carcinoma cells. J Biol Chem 269: 27595–27602
Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S, Rischin D, Eek R, Horai T, Noda K, Takata I, Smit E, Ayerbuch S, Macleod A, Feyereislova A, Dong RP, Baselga J (2003) Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer. J Clin Oncol 21: 2237–2246
Greenfield C, Hiles I, Waterfield MD, Federwish M, Wollmer A, Blundell L, McDonald N (1989) Epidermal growth factor binding induces a conformational change in the external domain of its receptor. EMBOJ 8: 4115–4123
Herbst RS, Hong WK (2002) IMC-C225, an anti-epidermal growth factor receptor monoclonal antibody for treatment of head and neck cancer. Semin Oncol 29: 18–30
Huang S, Armstrong EA, Benavente S, Chinnayan P, Harari P (2004) Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res 64: 5355–5362
Kowanetz K, Crosetto N, Haglund K, Schmidt MH, Hledin CH, Dikic I (2004) Suppressors of T-cell receptor signaling Sts-1 and Sts-2 bind to Cbl and inhibit endoyctosis of receptor tyrosine kinases. J Biol Chem 279: 32786–32795
Lynch TJ, Bell DW, Sordella R, Gurubhjagavatula S, Okimoto RA, Branningan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350: 2129–2139
Matar P, Rojo F, Cassia R, Moreno-Bueno G, Di Cosimo S, Tarbernero J, Guzman M, Rodriguez S, Arribas J, Palacios J, Baselga J (2004) Combined epidermal growth factor receptor targeting with the tyrosine kinase inhibitor gefitinib (ZD1839) and the monoclonal antibody cetuximab (IMC-C225): superiority single-agent receptor targeting. Clin Cancer Res 10: 6487–6501
Mendelsohn J (2002) Targeting the epidermal growth factor receptor for cancer therapy. J Clin Oncol 20: 1–13
Modi S, Seidman AD (2002) An update on epidermal growth factor receptor inhibitors. Curr Oncol Rep 4: 47–55
Noonberg SB, Benz CC (2000) Tyrosine kinase inhibitors targeted to the epidermal growth factor receptor subfamily: role as anticancer agents. Drugs 59: 753–767
Olivier S, Formento P, Fischel JL, Etienne MC, Milano G (1990) Epidermal growth factor receptor expression and suramin cytotoxicity in vitro. Eur J Cancer 26: 867–871
Opresko LK, Chang CP, Will BH, Burke PM, Gill GN, Wiley HS (1995) Endocytosis and lysosomal targeting of epidermal growth factor receptors are mediated by distinct sequences independent of the tyrosine kinase domain. J Biol Chem 270: 4325–4333
Prewett M, Rockwell P, Rockwell RF, Giorgo NA, Mendelsohn J, Scher HI, Goldstein NI (1996) The biologic effects of C225, a chimeric monoclonal antibody to the EGFR, on human prostate carcinoma. J Immunother Emphasis Tumor Immunol 19: 419–427
Ranson M, Mansoor W, Jayson G (2002) ZD1839 (Iressa): a selective EGFR-TK inhibitor. Expert Rev Anticancer Ther 2: 161–168
Saltz LB, Meropol NJ, Loehrer PJ, Needle MN, Kopit J, Mayer RJ (2004) Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol 22: 1201–1208
Thomas SM, Grandis JR (2004) Pharmacokinetic and pharmacodynamic properties of EGFR inhibitors under clinical investigation. Cancer Treat Rev 30: 255–268
Volm M, Kaufman M, Hinderer H, Goertler J (1970) Schnellmethode zur sensibilitätstestung malignen tumoren. Klin Wochenschr 48: 374–376
Waterman H, Yarden Y (2001) Molecular mechanisms underlying endocytosis and sorting of ErbB receptor tyrosine kinases. FEBS Lett 490: 142–152
Yarden Y (2001) The EGFR family and its ligands in human cancer: signalling mechanisms and therapeutic opportunities. Eur J Cancer 37: 3–8
Zwick E, Bange J, Ullrich A (2002) Receptor tyrosine kinases as targets for anticancer drugs. Mol Med 8: 17–23
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Fischel, JL., Formento, P. & Milano, G. Epidermal growth factor receptor double targeting by a tyrosine kinase inhibitor (Iressa) and a monoclonal antibody (Cetuximab). Impact on cell growth and molecular factors. Br J Cancer 92, 1063–1068 (2005). https://doi.org/10.1038/sj.bjc.6602428
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6602428
Keywords
This article is cited by
-
Combined effects of aspirin and vitamin D3 on two OSCC cell lines: a preliminary study
Biotechnology Letters (2018)
-
Autophagy Interplays with Apoptosis and Cell Cycle Regulation in the Growth Inhibiting Effect of Trisenox in HEP-2, a Laryngeal Squamous Cancer
Pathology & Oncology Research (2015)
-
Bortezomib overcomes MGMT-related resistance of glioblastoma cell lines to temozolomide in a schedule-dependent manner
Investigational New Drugs (2013)
-
Phosphorylated AKT and MAPK expression in primary tumours and in corresponding metastases and clinical outcome in colorectal cancer patients receiving irinotecan-cetuximab
Journal of Translational Medicine (2012)
-
Efficient Blockade of Akt signaling is a determinant factor to overcome resistance to Matuzumab
Molecular Cancer (2011)
