
Abstract
Reactive oxygen species (ROS) produced by tumor necrosis factor-α (TNF-α) have an important function in cell death by activating c-Jun N-terminal kinase. However, the exact mechanism of mitochondrial ROS production, after TNF-α stimulation, is not clearly understood. In this study, we determined that ROS modulator 1 (Romo1) and B-cell lymphoma-extra large (Bcl-XL) are directly associated with TNF-α-induced ROS production. In response to TNF-α, TNF complex II, which consists of receptor-interacting protein 1, TNF receptor-associated protein with death domain, TNF receptor-associated factor 2, Fas-associated death domain protein, and pro-caspase-8, binds to the C-terminus of Romo1 located in the mitochondria. Concurrently, Romo1 recruits Bcl-XL to reduce the mitochondrial membrane potential, resulting in ROS production and apoptotic cell death. On the basis of these results, we suggest that Romo1 is a molecular bridge between TNF-α signaling and the mitochondria for ROS production that triggers TNF-α-mediated apoptosis, as well as a novel target in the development of anti-inflammatory agents that block the origin of ROS production.
Similar content being viewed by others


Main
Tumor necrosis factor-α (TNF-α) is a major mediator of inflammation. There are two main pathways in TNF-α signaling. One is the pro-survival/pro-inflammatory pathway that activates NF-κB and MAPK through TNF-α-induced signaling complex I.1 After binding of the trimeric TNF-α to the cell surface receptor TNF–R1, the silencer of death domain dissociates from TNF–R1 complexes, and the TNF receptor-associated protein with death domain (TRADD) – an adaptor protein – binds to the cytoplasmic domain of the receptor.2 TRADD recruits additional proteins including TNF receptor-associated factor 2 (TRAF2), receptor-interacting protein 1 (RIP1), and cellular inhibitor of apoptosis proteins to form TNF-α-induced signaling complex I.3, 4 The other TNF-α signaling pathway is the proapoptotic pathway through TNF-α-induced signaling complex II, in which reactive oxygen species (ROS), a caspase cascade, and the mitochondria function as downstream mediators. If TRADD, TRAF2, and RIP1 dissociate from the receptor, they recruit Fas-associated death domain protein (FADD) and pro-caspase-8 to form TNF-α-induced signaling complex II, which is implicated in signaling apoptosis.5, 6 This TNF-α-induced cell death pathway is normally blocked by concomitant activation of NF-κB.
ROS generation, after TNF-α binding, has been reported to be involved in both cell survival and cell death, and the main source of ROS generation that contributes to TNF-α-induced cell death is the mitochondrion.7, 8, 9 However, another study reported that NADPH oxidase is the source of ROS generation after TNF-α treatment.10 ROS are known to contribute to cell death by inducing mitochondrial membrane permeabilization and sustaining c-Jun N-terminal kinase (JNK) activation.11, 12 JNK is activated by the activation of apoptosis signal-regulating kinase-1 or by ROS-mediated inactivation of MKPs.13, 14 There are several reports regarding the mechanism of JNK-induced apoptosis in response to TNF-α. JNK induces cFLIP degradation through Itch activation to trigger apoptosis.15, 16, 17 JNK also contributes to TNF-α-stimulated cell death by cleavage of Bid into jBid, which stimulates the release of Smac/Diablo into the cytosol to activate caspases.18
B-cell lymphoma-extra large (Bcl-XL) is known to stabilize Δψm to maintain mitochondrial homeostasis.19, 20 Bcl-XL-promoted Δψm stabilization seems to prevent cell death because massive cell death was observed in Bcl-XL-deficient mice.21 TNF-α/D-GalN-induced cell death from a variety of stimuli was blocked by Bcl-XL overexpression.22, 23 Gottlieb et al.24 suggested that TNF-α treatment induces an early decrease in Δψm and subsequent ROS formation, which promotes a late decrease in Δψm, and that Bcl-XL expression inhibits the early decrease in Δψm and the subsequent ROS generation after TNF-α stimulation.
ROS modulator 1 (Romo1) is localized in the mitochondria and induces mitochondrial ROS production through complex III of the mitochondrial electron transport chain.25, 26, 27 The ROS originating from the endogenous Romo1 protein appear to be indispensable for both normal and cancer cell proliferation, because Romo1 knockdown inhibits cell-cycle transition through inhibition of Erk activation and p27Kip1 expression.28, 29 Romo1 expression is induced by an external stimulus such as 5-FU, and it is upregulated in senescent cells and in most cancer cells.25, 26, 30 As the mitochondrial origin of ROS formation triggered by TNF-α is not clear, and Romo1 contributes to ROS production in the mitochondria, we investigated whether TNF-α-induced ROS are associated with Romo1.
Results
Romo1 is responsible for TNF-α-induced ROS production
We examined TNF-α-induced ROS production by staining cells with the oxidant-sensitive dye, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA), after Romo1 knockdown by Romo1 small-interfering RNA (siRNA) transfection. As ROS produced by TNF-α are eliminated by FHC and MnSOD, which are products of TNF-α-activated NF-κB signaling,31 cells were treated with the translation inhibitor, cycloheximide (CHX), to suppress NF-κB signaling stimulated by TNF-α. As expected, TNF-α treatment increased ROS levels in cells pretreated with CHX (Figure 1a). Interestingly, the TNF-α-stimulated ROS increase was completely blocked by Romo1 knockdown. The decrease in ROS formation was quantified using MetaMorph software and is illustrated in HeLa cells (Figure 1a) and human embryo kidney (HEK) 293 cells (Supplementary Figure S1a). Butylated hydroxy anisol (BHA), an antioxidant, was also added to cells pretreated with CHX. The ROS level in these cells was downregulated by Romo1 knockdown itself (Figure 1), which is consistent with earlier reports.28, 29 This experiment was also carried out again in the cells treated with H2O2 as a positive control. HeLa cells were transfected with 100 nM of control siRNA or Romo1 siRNA for 16 h and treated with H2O2 (100 μM) for 1 h. As expected, ROS increase was observed in the cells transfected with control siRNA (Supplementary Figure S1b). Interestingly, ROS production induced by H2O2 treatment was blocked in the cells transfected with Romo1 siRNA. It has been reported that ROS induces ROS release.32 Therefore, we suggest that H2O2 treatment induces ROS production through Romo1. The exact mechanism as to how Romo1 may be involved in H2O2-triggered ROS induction is now under investigation. Suppression of a TNF-α-induced ROS increase by Romo1 knockdown was also observed by flow cytometry, and the same result was obtained in HeLa cells (Figure 1b) and HEK 293 cells (Supplementary Figure S1c). As dominant negative-I-κB (DN-I-κB) overexpression inhibits NF-κB translocation to the nucleus, DN-I-κB was transfected into HeLa cells to inhibit the TNF-α-induced NF-κB activation. Blockage of TNF-α-stimulated nuclear translocation of NF-κB on DN-I-κB expression was observed by immunofluorescence analysis (Figure 1c). TNF-α treatment also increased ROS levels in cells transfected with DN-I-κB, and the ROS increase was completely suppressed by Romo1 knockdown in HeLa cells (Figure 1d) and HEK 293 cells (Supplementary Figure S1d). The inhibition of ROS formation was quantified by MetaMorph software and is shown in Figure 1e and Supplementary Figure 1e. Suppression of TNF-α-induced ROS formation by Romo1 knockdown was also observed by flow cytometry (Figure 1f). To measure the transfection efficiency of Romo1 siRNA by flow cytometry, Romo1 siRNA-FITC was transfected into these two cell lines; the Romo1 siRNA transfection efficiencies were above 90% (data not shown). Romo1 knockdown was examined by western blotting (Figure 1g) or RT-PCR (Supplementary Figure S1f), and the result was consistent with earlier reports.26, 27, 28, 29

TNF-α-induced ROS production is blocked by Romo1 siRNA transfection. (a) After transfection of Romo1 siRNA (100 nM) for 16 h, HeLa cells were incubated with TNF-α (20 ng/ml) and CHX (10 μg/ml) in the presence or absence of BHA (100 μM) for the indicated times. The cells were stained with CM-H2DCFDA (5 μg/ml) for 30 min and then observed by confocal microscopy. For quantification purposes, the computer overlaid the images obtained from fluorescence microscopy, and CM-H2DCFDA fluorescence was analyzed with MetaMorph software (Universal Imaging). The arrow indicates apoptotic bodies. The results represent the means (±S.E.) of three independent experiments performed in triplicate. *P<0.05; **P<0.01; ***P<0.001 versus control siRNA by two-way ANOVA. (b) After transfection of Romo1 siRNA for 16 h, HeLa cells were incubated with TNF-α and CHX, in the presence or absence of BHA, for the indicated times, then stained with CM-H2DCFDA for 30 min, and analyzed by flow cytometry. (c) The subcellular localization of NF-κB was examined in HeLa cells transfected with DN-I-κB. NF-κB translocation into the nucleus was inhibited in cells expressing DN-I-κB (arrow). (d) TNF-α-induced ROS production was measured in HeLa cells transfected with DN-I-κB. The cells were stained with CM-H2DCFDA for 30 min and then observed by fluorescence microscopy. (e) CM-H2DCFDA fluorescence in HeLa was analyzed with MetaMorph software. The results represent the means (±S.E.) of three independent experiments performed in triplicate. ***P<0.001 versus control siRNA by two-way ANOVA. (f) After transfection of DN-I-κB, HeLa cells were treated with TNF-α, stained with CM-H2DCFDA, and then analyzed by flow cytometry. (g) HeLa cells were transfected with Romo1 siRNA and harvested after the indicated times. Romo1 knockdown was examined by western blotting. β-actin was used as a loading control. Bar: 50 μm
TNF-α-induced JNK activation and cell death are regulated by Romo1-mediated ROS production
We examined whether Romo1 regulated TNF-α-triggered JNK activation and cell death. As shown in Figure 2a, TNF-α treatment induced prolonged JNK activation in cells pretreated with CHX, and Romo1 knockdown inhibited this JNK activation. Next, we examined whether Romo1 was involved in TNF-α-induced apoptotic cell death. Romo1 siRNA was transfected into HeLa cells, and cell death was observed after TNF-α treatment. Figure 2b and Supplementary Figure S2a show that TNF-α stimulation in the presence of CHX induced cell death, and that cell death was inhibited by Romo1 knockdown or BHA treatment. However, Romo1 siRNA transfection failed to completely inhibit cell death, showing that another signaling pathway independent of Romo1 also exists in TNF-α-stimulated cell death. To observe apoptotic body formation in TNF-α-induced cell death, we stained cells with DAPI and visualized them by fluorescence microscopy after Romo1 knockdown. As shown in Figure 2c and Supplementary Figure S2b, TNF-α treatment increased the formation of apoptotic bodies, whereas Romo1 knockdown decreased their formation. To see the cleavage of mediators involved in TNF-α-induced apoptosis, western blotting was performed after Romo1 knockdown. Figure 2d shows that cleavage of caspase-8, caspase-3, PARP, RIP1, and Bid was observed after TNF-α treatment, and that Romo1 knockdown inhibited these cleavages, showing that Romo1 is an upstream mediator of caspase-8 and Bid in TNF-α-triggered apoptosis. Next, cells were treated with zVAD, a caspase inhibitor, before TNF-α treatment to determine whether zVAD blocked TNF-α-induced cell death. As expected, Romo1 knockdown partially inhibited TNF-α-induced cell death, whereas zVAD treatment more effectively prevented cell death (Figure 2e; Supplementary Figure S2c). These results show that Romo1 is an important mediator of cell death triggered by TNF-α.

TNF-α-induced cell death is suppressed by Romo1 knockdown. (a) After transfection of Romo1 siRNA into HeLa cells for 16 h, cells were incubated with TNF-α and CHX for the indicated times. Western blotting was performed using antibodies to the indicated proteins. β-actin was used as a loading control. 15′, 15 min; 30′, 30 min. (b) Cells were incubated with TNF-α and CHX for the indicated times, and the cells were counted by staining with trypan-blue. The results represent the means (±S.E.) of three independent experiments performed in triplicate. ***P<0.001 versus control siRNA by two-way ANOVA. (c) After TNF-α treatment with CHX for the indicated times, HeLa cells were stained with DAPI, and apoptotic bodies were observed by fluorescence microscopy. The number of cells exhibiting apoptotic death was counted out of a total of 200 cells. The data represent the mean ± SD of three determinations. **P<0.01; ***P<0.001 versus control siRNA by two-way ANOVA. (d) After transfection of Romo1 siRNA into HeLa cells for 16 h, the cells were incubated with TNF-α and CHX for the indicated times. Western blotting was performed using antibodies to the indicated proteins. (e) Cells were incubated with TNF-α and CHX for 6 h in the presence of zVAD, and the cells were counted by staining with trypan blue. The results represent the means (±S.E.) of three independent experiments performed in triplicate. #P<0.05, ***P<0.001 versus zero time of each case by two-way ANOVA
TNF-α-induced signaling complex II binds to the C-terminal region of Romo1 in response to TNF-α
To see the interaction between endogenous Romo1 and TNF complex II, RIP1 was immunoprecipitated with anti-RIP1 antibody, and western blotting was performed with an anti-Romo1 antibody. Figure 3a shows that RIP1 interacted with endogenous Romo1 at 15 min and continued to 4 h during TNF-α stimulation. To confirm this interaction, Flag-tagged Romo1 was transfected into HEK 293 cells, and the Romo1 protein was immunoprecipitated with Flag antibody to perform western blotting with RIP1, TRADD, TRAF-2, FADD, and caspase-8. Figure 3b shows that Romo1 interacted with RIP1 as well as TRADD, TRAF-2, FADD, and caspase-8 at 15 min and continued to 4 h during TNF-α stimulation. The colocalization of Romo1 and RIP1 in HEK 293 cells was also observed by confocal microscopy. As shown in Figure 3c, on TNF-α stimulation, the green Flag-Romo1 signals completely overlapped with the red RIP1 signals. The green RIP1 signals also overlapped with the mitochondria-specific MitoTracker red fluorescent dye (Figure 3d). In addition, we examined the colocalization of the other TNF-α signaling complex II proteins with Romo1; Supplementary Figure S3 shows that they translocated into the mitochondria to interact with Romo1.


TNF-α stimulation induces interaction of TNF-α-induced signaling complex II with the C-terminal domain of Romo1. (a) After HEK 293 cells were incubated with TNF-α and CHX, RIP1 was immunoprecipitated with anti-RIP1 antibody, and western blotting was performed with anti-Romo1 antibody and the indicated antibodies. (b) After HEK 293 cells were transfected with Flag-tagged Romo1, cells were incubated with TNF-α and CHX. The lysates were subjected to immunoprecipitation (IP) with anti-Flag antibody. The precipitates were analyzed by western blotting with the indicated antibodies. Ig, immunoglobulin. (c) RIP1 was colocalized with Romo1. HEK 293 cells were transfected with GFP-tagged Romo1 (green) and visualized by confocal microscopy using anti-RIP1 antibody (red). (d) RIP1 was translocated into mitochondria after TNF-α stimulation. After HeLa cells were incubated with TNF-α, the cells were treated with anti-RIP1 antibody (green) and MitoTracker (red). The colocalization of RIP1 and mitochondria was visualized by confocal microscopy. (e) Schematic diagram showing the structure of Romo1. Romo1 deletion mutants were constructed by PCR. (f) HEK 293 cells were transfected with Flag-tagged Romo1 deletion mutants. After the cells were incubated with TNF-α and CHX, lysates were subjected to immunoprecipitation with an anti-Flag antibody. The precipitates were analyzed with the indicated antibodies. (g, h) After HeLa cells were transfected with Flag-tagged Romo1 deletion mutants, their localization (g) or ROS production (h) was observed by confocal microscopy. The cells were treated with anti-Flag antibody (g, green; h, red) or MitoTracker (g, red). ROS production was stained with CM-H2DCFDA (h, green). ROS production was also observed by fluorescence microscopy and quantified by MetaMorph software. Bars: (c, d, h) 25 μm; (g) 50 μm
In an earlier report, we suggested that Romo1 is a transmembrane protein with a molecular weight of approximately 8.9 kDa.25 The transmembrane domain was also predicted using SMART (http://smart.embl-heidelberg.de) and CBS Prediction Servers (http://www.cbs.dtu.dk/services/TMHMM). Therefore, we made two deletion mutants of Romo1, designated Romo1-ΔC and Romo1-ΔN, and examined which domain interacted with TNF-α-induced signaling complex II (Figure 3e). As shown in Figure 3f, the C-terminal region of Romo1 interacted with TNF-α-induced signaling complex II, showing that the C-terminal domain of Romo1 was oriented into the cytosol. Next, we observed whether these deletion mutants localized to the mitochondria and induced mitochondrial ROS generation. Figure 3g and h shows that only the N-terminal deletion mutant localized to the mitochondria and enhanced ROS levels. In contrast, the C-terminal deletion mutant was found in the whole cell and failed to generate mitochondrial ROS. These results show that the mitochondrial localization signal is located in the C-terminal domain and that the C-terminal domain is important for TNF-α-induced ROS production. To test the Romo1 topology, the mitochondria were isolated from HEK 293 cells transfected with N-terminally Flag-tagged Romo1 or C-terminally myc-tagged Romo1. After digestion with proteinase K in the presence or absence of Triton X-100, western blotting was performed using anti-Flag or myc antibodies. As shown in Figure 4, the C-terminally myc-tagged Romo1 was digested by proteinase K. However, the N-terminally Flag-tagged Romo1 protein remained. Instead, the N-terminally Flag-tagged Romo1 was detected at a smaller size. This result shows that the C-terminus of Romo1 is exposed to the cytosol. Voltage-dependent anion channel (VDAC), in the outer mitochondrial membrane, and Mn-SOD, in the mitochondrial matrix, were used as positive controls.

Romo1 topology on the mitochondrial membrane. Intact mitochondria were isolated from HEK 293 cells transfected with Flag-Romo1 or Romo1-myc and digested with 50 μg/ml proteinase K in the absence or presence of 1% Triton X-100. Western blotting was performed using antibodies to the indicated proteins. *, nonspecific band
TNF-α-induced initial decrease of Δψm through Romo1 is followed by ROS production
TNF-α induces the initial decrease of Δψm, leading to ROS generation and subsequent loss of Δψm, which triggers ROS formation to induce apoptosis.24, 33 To examine whether TNF-α-induced loss of Δψm is regulated by Romo1, HeLa cells were transfected with Romo1 siRNA for 16 h, pretreated with CHX, BHA, stigmatellin, or rotenone, and then incubated with TNF-α for the indicated times; Δψm decrease was then determined by staining the cells with 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1). Red fluorescence indicates a high Δψm and green fluorescence conversely indicates a low Δψm. Romo1 knockdown itself stabilized Δψm, showing that Romo1 expression contributed to loss of Δψm (Figure 5a and b). After TNF-α treatment, the decrease of Δψm was detected by fluorescence microscopy in cells transfected with control siRNA; this decrease of Δψm was inhibited by Romo1 knockdown or BHA treatment (Figure 5a and b). Interestingly, although the TNF-α-induced decrease in Δψm was inhibited by Romo1 knockdown at 15 min, the TNF-α-induced decrease of Δψm was not blocked by BHA treatment (Figure 5a and b). The earlier report showed that Romo1-mediated ROS production was generated from the mitochondrial electron transport chain.26 Therefore, we examined whether the inhibitors of the mitochondrial electron transport chain blocked the initial decrease of Δψm after TNF-α stimulation. Stigmatellin or rotenone also failed to inhibit the initial TNF-α-induced decrease of Δψm (Figure 5a and b). These results indicate that the initial TNF-α-induced decrease of Δψm (at 15 min) was only blocked by Romo1 knockdown, and that the initial decrease of Δψm mediated by Romo1 after TNF-α stimulation was independent of ROS production. As shown in Figure 5b, the decrease of Δψm – indicated by the downshift of the dotted squares – was accelerated in control cells at 4 h, and BHA or stigmatellin treatment inhibited the late TNF-α-induced decrease of Δψm (at 4 h). Figure 5c and d shows the quantified data of Figure 5b by representing the ratio of the signal of high Δψm (red) to the signal of low Δψm (green). As expected, rotenone treatment failed to block the late TNF-α-induced decrease of Δψm, showing that the late TNF-α-induced decrease of Δψm was triggered by ROS originating from complex III in the mitochondrial electron transport chain. Without CHX treatment, the late TNF-α-induced decrease of Δψm was recovered, because antioxidant enzymes such as FHC and MnSOD induced by NF-κB seemed to eliminate ROS generated from mitochondria (Figure 5e). To confirm these results, Δψm decrease was also examined by staining the cells with tetramethylrhodamine methyl ester (TMRM). The same result was obtained in the cells stained with TMRM, compared with the cells with JC-1 (Figure 5f). The change of Δψm was quantified using MetaMorph software and is illustrated in Figure 5g. On the basis of these results, we suggest that Romo1 mediates the early TNF-α-induced decrease of Δψm to trigger ROS production, which leads to the late decrease of Δψm and subsequent ROS accumulation.


TNF-α stimulation induces a decrease in Δψm through Romo1 before ROS generation. (a, b) After transfection of Romo1 siRNA for 16 h, HeLa cells were incubated with TNF-α in the presence of BHA, stigmatellin (1 μM), or rotenone (1 μM) for the indicated times, and the loss of Δψm was determined by fluorescence microscopy (a) or flow cytometry (b) by staining with JC-1 (5 μM) for 30 min. Cells were incubated with CHX, BHA, stigmatellin, or rotenone for 30 min before TNF-α treatment. The downshift of dots (b, a dotted square) indicates a decrease in Δψm. Red JC-1 aggregates indicating high Δψm are detectable in the FL2 channel, and green JC-1 monomers indicating low Δψm are detectable in the FITC channel (FL1). (c) HeLa cells were transfected with Romo1 siRNA, and the loss of Δψm was determined by flow cytometry by staining with JC-1. Δψm was represented as the ratio of the signal of high Δψm (red) to the signal of low Δψm (green). The results represent the means (±S.E.) of three independent experiments performed in triplicate. *P<0.05; **P<0.01 versus control siRNA by one-way ANOVA. (d, e) After transfection of Romo1 siRNA for 16 h, HeLa cells were incubated with TNF-α in the presence (d) or absence (e) of CHX for the indicated times, and then the loss of Δψm was determined by flow cytometry. ##P<0.01; ***, ###, ¥¥¥, §§§P<0.001 versus zero time of each case by two-way ANOVA. (f) After transfection of Romo1 siRNA for 16 h, HeLa cells were incubated with TNF-α in the presence or absence of CHX with BHA, stigmatellin (1 μM), or rotenone (1 μM) for the indicated times, and the loss of Δψm was determined by fluorescence microscopy by staining with TMRM. (g) Red fluorescence was also observed by fluorescence microscopy and quantified by MetaMorph software. **,##, ¥¥, §§P<0.01; ***P<0.001 versus zero time of each case by two-way ANOVA. Bar: 50 μm
Romo1 regulates TNF-α-induced early decrease of Δψm through interaction with Bcl-XL
First, we examined whether endogenous Romo1 interacted with the Bcl-XL protein through coimmunoprecipitation assays. To examine this interaction, Bcl-XL was immunoprecipitated with its antibody, and western blotting was performed with anti-Romo1 antibody. As shown in Figure 6a, Bcl-XL was associated with Romo1, but not with TNF-α-induced signaling complex II components. Interestingly, Bax was released from Bcl-XL after TNF-α stimulation, and Romo1 showed weak binding with Bcl-XL without TNF-α stimulation (Figure 6a). To confirm this interaction, Flag-tagged Romo1 was transfected into HEK 293 cells, and the Romo1 protein was immunoprecipitated with Flag antibody to perform western blotting with Bcl-XL or Bcl-2 antibodies. On TNF-α stimulation, concurrent Romo1 binding to Bcl-XL, compared with the binding of Romo1–TNF-α-induced signaling complex II, was observed; the binding was increased for up to 4 h. In contrast, Romo1 did not associate with Bcl-2, Bax, or VDAC (Figure 6b). Using deletion mutants of Romo1, we explored which region of Romo1 was required for binding to Bcl-XL. As shown in Figure 6c, the C-terminal region of Romo1 was necessary for binding to Bcl-XL.

TNF-α stimulation induces interaction of Bcl-XL with the C-terminal domain of Romo1. (a) After HEK 293 cells were incubated with TNF-α and CHX, Bcl-XL was immunoprecipitated with anti-Bcl-XL antibody, and western blotting was performed with anti-Romo1 antibody and indicated antibodies. (b) After HEK 293 cells were transfected with Flag-Romo1, cells were incubated with TNF-α and CHX. The lysates were subjected to immunoprecipitation with anti-Flag antibody. The precipitates were analyzed by western blotting with the indicated antibodies. (c) After HEK 293 cells were transfected with Romo1 deletion mutants, the cells were incubated with TNF-α and CHX. The lysates were subjected to immunoprecipitation with anti-Bcl-XL antibody. The precipitates were analyzed by western blotting with the indicated antibodies
In this study, we showed an early TNF-α-induced decrease of Δψm through Romo1 and the interaction of Romo1 with Bcl-XL. Therefore, we examined whether the early TNF-α-induced decrease of Δψm mediated by Romo1 was regulated by Bcl-XL. As shown in Figure 7a, the downshift of dots was increased in cells transfected with Romo1 and inhibited by Bcl-XL expression. Figure 7b shows the quantified data of Figure 7a by representing the ratio of the signal of high Δψm (red) to the signal of low Δψm (green). Next, we examined whether Bcl-XL prevented the early TNF-α-induced decrease of Δψm (at 15 min). As shown in Figure 7c–e, the early TNF-α-induced decrease of Δψm was blocked by Bcl-XL expression. From these results, we suggest that Romo1 mediated the early TNF-α-induced decrease of Δψm by binding to Bcl-XL and preventing its function.

Bcl-XL expression suppresses the early TNF-α-induced decrease in Δψm mediated by Romo1. (a, b) After HeLa cells were transfected with Romo1 or Bcl-XL, the loss of Δψm was determined by flow cytometry by staining with JC-1. The downshift of dots (a dotted square) indicates the decrease in Δψm (a). Δψm was represented as the ratio of the signal of high Δψm (red) to the signal of low Δψm (green) (b). The results represent the means (±S.E.) of three independent experiments performed in triplicate. *P<0.05; **P<0.01 versus vector by one-way ANOVA. (c, d, e) After HeLa cells were transfected with Romo1, Bcl-XL, or Romo1 and Bcl-XL, cells were treated with TNF-α in the presence of CHX for the indicated times. The loss of Δψm was determined by flow cytometry by staining with JC-1. Red JC-1 aggregates indicating that high Δψm are detectable in the FL2 channel, and green JC-1 monomers indicating low Δψm are detectable in the FITC channel (FL1). The Δψm was represented as the ratio of the signal of high Δψm (red) to the signal of low Δψm (green). The results represent the means (±S.E.) of three independent experiments performed in triplicate. **, ¥¥P<0.01; ###P<0.001 versus zero time of each case by two-way ANOVA (the color reproduction of this figure is available on the html full text version of the manuscript)
Bcl-XL prevents TNF-α-induced cell death mediated by Romo1
TNF-α-induced cell death is known to be suppressed by Bcl-XL expression.24 We also investigated whether Bcl-XL prevented TNF-α-induced ROS production; this production was measured by flow cytometry after Bcl-XL transfection. As shown in Figure 8a and b, Bcl-XL overexpression blocked TNF-α-induced ROS formation. Next, we investigated whether Bcl-XL prevented Romo1-induced cell death. After Bcl-XL and Romo1 were transfected into HeLa cells, the number of cells was counted. As shown in Figure 8c, Bcl-XL prevented Romo1-induced cell death. To address whether TNF-α-induced cell death mediated by Romo1 was regulated by Bcl-XL, HeLa cells were transfected with Romo1, Bcl-XL, or Romo1 and Bcl-XL. As shown in Figure 8d, Bcl-XL effectively inhibited not only TNF-α-induced cell death, but also TNF-α-induced cell death enhanced by Romo1 expression. In Figure 6c, we showed that the C-terminal region of Romo1 was important for binding to Bcl-XL. Therefore, HeLa cells were transfected with the C-terminal region of Romo1 or the N-terminal region of Romo1. The C-terminal region of Romo1, as well as the full-length version, did induce cell death (Figure 8e; Supplementary Figure S4). Unexpectedly, the N-terminal region of Romo1 also induced cell death. We do not know why the N-terminal region of Romo1 also induced cell death, and this requires further investigation. However, Bcl-XL expression effectively recovered the cell death enhanced by the C-terminal region of Romo1, but not the cell death enhanced by the N-terminal region of Romo1.

Bcl-XL expression suppresses TNF-α-induced ROS production and cell death mediated by Romo1. (a, b) After HeLa cells were transfected with Bcl-XL for 36 h, cells were incubated with TNF-α and CHX for the indicated times. The cells were stained with CM-H2DCFDA for 30 min, and ROS generation was analyzed by flow cytometry. *P<0.05; ***P<0.001 versus vector by two-way ANOVA. Solid line, not treated with TNF-α plus CHX; Dotted line, treated with TNF-α plus CHX. (c) After HeLa cells were transfected with Romo1, Bcl-XL, or Romo1 and Bcl-XL for 36 h, the cells were observed under an optical microscope (top). The cells were counted by staining with trypan blue (bottom). The results represent the means (±S.E.) of three independent experiments performed in triplicate. **, ##P<0.01; ###P<0.001 by two-way ANOVA. (d) After HeLa cells were transfected with Romo1, Bcl-XL, or Romo1 and Bcl-XL, the cells were treated with TNF-α in the presence of CHX for the indicated times. The cells were observed under an optical microscope (top) and counted by staining with trypan blue (bottom). *P<0.05; **P<0.01 versus vector by two-way ANOVA. (e) After HeLa cells were transfected with Romo1 deletion mutants for 36 h, the cells were counted by staining with trypan blue. The results represent the means (±S.E.) of three independent experiments performed in triplicate. **, ##, ¥¥P<0.01 versus zero time of each case by two-way ANOVA. Bar: 50 μm (the color reproduction of this figure is available on the html full text version of the manuscript)
Discussion
ROS have an important function in cell survival and cell death triggered by TNF-α signaling, and the main source in ROS generation required for TNF-α-induced cell death is the mitochondria.8, 34, 35, 36 Therefore, we were curious whether Romo1 was involved in TNF-α-induced ROS production, because Romo1 is located in the mitochondrial membrane and induces ROS generation.25 As shown in Figure 1, TNF-α-triggered ROS production was completely blocked by Romo1 knockdown, and the same result was obtained with BHA treatment. This result shows that Romo1 is responsible for TNF-α-induced ROS production, and that Romo1 should be located in the outer membrane of the mitochondria to sense the signal elicited by TNF-α. In our earlier report, we showed that the molecular weight of Romo1 is approximately 8.9 kDa, and that Romo1 is a transmembrane protein with a single transmembrane domain.25 In this study, we showed that Romo1 is associated with TNF complex II, which consists of RIP1, TRADD, TRAF2, FADD, and pro-caspase-8, in response to TNF-α stimulation, and that the C-terminus of Romo1 interacts with TNF-α-induced signaling complex II (Figure 3). From the results obtained in this study, we suggest that TNF-α-induced signaling complex II interacts with the C-terminus of Romo1, which is oriented into the cytosol, and transmits the TNF-α signal to mitochondria. It is unlikely that Romo1 generates ROS in response to TNF-α signaling similar to NADPH oxidase and other enzymes such as xanthine oxidase, cyclooxygenases, and lipoxygenases. Romo1 consists of 79 amino acids and has no homology domain when compared with these enzymes. It seems that Romo1, which is located in the mitochondrial outer membrane, functions as a modulator to supply ROS from complex III of the mitochondrial respiratory chain into the cytosol when the TNF-α signal is delivered to mitochondria through TNF-α-induced signaling complex II.
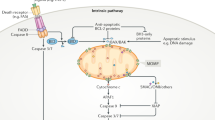
How does Romo1 modulate ROS release from mitochondria? Our suggestion is that Romo1 transmits the TNF-α-triggered signal to mitochondria by forming a complex with Bcl-XL to decrease Δψm, resulting in ROS production. Bcl-XL has been reported to stabilize mitochondrial membrane potential (Δψm) to maintain mitochondrial homeostasis.19, 20 An early event in Romo1-mediated ROS production is the concurrent binding of the C-terminus of Romo1 to Bcl-XL, compared with Romo1 binding to TNF-α-induced signaling complex II (Figure 6). Interestingly, Bcl-XL binding to TNF-α-induced signaling complex II is not observed in response to TNF-α. This result indicates that TNF-α-induced signaling complex II-Romo1 binding and Romo1–Bcl-XL interaction occurred simultaneously, and that TNF-α-induced signaling complex II-Romo1 binding led to Romo1–Bcl-XL interaction. We do not know the exact mechanism by which TNF-α-induced signaling complex II-Romo1 induces Romo1–Bcl-XL interaction, and this remains to be clarified through further study. The second event is the loss of Δψm resulting from Romo1–Bcl-XL interaction, which triggers ROS formation. In this study, the TNF-α-induced decrease of early Δψm was inhibited by Romo1 knockdown; however, it was not blocked by BHA or mitochondrial complex inhibitors (Figure 5), showing that the initial TNF-α-induced decrease of Δψm is an early event before ROS induction. Our results also showed that Bcl-XL overexpression suppressed these events, which were triggered by TNF-α stimulation (Figure 7). Thus, Romo1 interacts with Bcl-XL to decrease Δψm, leading to subsequent ROS generation after TNF-α stimulation. Interestingly, a basal level of interaction between Romo1 and Bcl-XL was observed without TNF-α treatment (Figure 6). Romo1 overexpression without TNF-α treatment, therefore, induces the loss of Δψm and enhances ROS levels. We showed that Romo1 overexpression caused the loss of Δψm in this study (Figure 7b), whereas ROS levels increased by Romo1 overexpression in an earlier report.25 These findings show that the basal interaction between Romo1 and Bcl-xL is important for ROS supply into the cytosol even without stimuli. Consistent with this suggestion, we showed that Romo1 knockdown decreased ROS levels in both normal and tumor cells.28, 29 Taken together, Romo1 is an important modulator to supply ROS into the cytosol. Without any signal, Romo1 supplies the minimal level of ROS required for cell survival and proliferation through the basal binding of Romo1 to Bcl-XL. If the cell needs enhanced ROS levels in response to a signal such as TNF-α, Romo1 recruits more Bcl-XL to increase ROS levels. The proposed TNF-α-induced ROS signaling pathway mediated by Romo1 is shown in Figure 9.

A proposed model for TNF-α-induced ROS production through Romo1 and Bcl-XL
Cell life or death, after TNF-α stimulation, is well controlled by the crosstalk between the pro-survival pathway and the proapoptotic pathway. Upregulated production of TNF-α or inappropriate activation of TNF-α signaling has been reported to contribute to a variety of diseases.3 TNF-α-induced cell death can be initiated by the mitochondria-dependent pathway (intrinsic pathway) or by the mitochondria-independent pathway (extrinsic pathway).37 In this study, we showed that Romo1 mediated TNF-α-induced ROS generation by forming a complex with TNF-α-induced signaling complex II and with Bcl-XL. However, Romo1 knockdown with Romo1 siRNA could not completely inhibit TNF-α-induced apoptotic cell death (Figure 2). In the extrinsic pathway, death receptor recruits pro-caspase-8 in TNF-α-induced signaling complex II and activates the caspase cascade to induce apoptotic cell death. Therefore, we assume that TNF-α-induced cell death in the presence of CHX can occur through both pathways, and that Romo1 knockdown inhibits only the mitochondria-dependent cell death pathway.
Materials and Methods
Cell culture and reagents
HEK 293 cells and HeLa cervix carcinoma cells were cultured in Dulbecco's modified Eagle's media (Gibco-Invitrogen, Grand Island, NY, USA) supplemented with 10% heat-inactivated FBS (Gibco-Invitrogen), sodium bicarbonate (2 mg/ml; Sigma-Aldrich, St. Louis, MO, USA), penicillin (100 units/ml), and streptomycin (100 μg/ml; Gibco-Invitrogen) and grown in 5% CO2 at 37°C. Human recombinant TNF-α was obtained from R&D System. zVAD, BHA, stigmatellin, rotenone, and TMRM were purchased from Sigma-Aldrich. CM-H2DCFDA, MitoSOX, and JC-1 were obtained from Molecular Probes (Eugene, OR, USA).
Antibodies
Mouse anti-TRADD, rabbit polyclonal anti-Bcl-XL, anti-TRAF2, anti-Mn-SOD, and anti-mitochondria-specific VDAC antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit polyclonal anti-caspase-3, anti-PARP, anti-phospho-JNK, anti-JNK, anti-phospho-c-Jun, anti-c-Jun, anti-Bid, anti-Bcl-2, and mouse anti-FADD antibodies were from Cell Signaling Technology (Danvers, MA, USA). Mouse anti-cytosol-specific-β-actin and anti-FLAG (M2) antibodies were from Sigma-Aldrich. Mouse anti-RIP antibody was from Transduction Lab (Lexington, KY, USA). Mouse anti-Myc antibody was from Invitrogen (Carlsbad, CA, USA). Mouse monoclonal antibody (mAb) against Romo1 was raised against the peptide sequences [YGQSQPSCFDRVK (corresponding to amino-acids 8–20 of Romo1) and RIGMRGRELMGGI (corresponding to amino-acids 44–56 of Romo1)]. A hybridoma cell line was supplied from Aprogen (Taejon, Republic of Korea) by the mAb production service. IgG was purified from hybridoma culture using protein G-agarose (Sigma-Aldrich).
siRNA transfections
Bioneer (Taejon, Republic of Korea) synthesized double-stranded siRNA oligonucleotides targeting Romo1.30 The cells (1 × 105) were seeded into six-well plates and then transfected with Romo1 siRNA using Lipofectamine (Gibco-Invitrogen) in cells at 70% confluency according to the manufacturer's instructions.
Plasmids
PCR amplification was performed to generate the Romo1 mutants using the following primers containing the BamHI or EcoRI restriction enzyme site: forward, 5′-GATCGGATCCCGCCGCCACCATGCCGGTGGCCGTG -3′ and reverse, 5′-GATCGAATTCCTTTACATTCCGATCCTGAGACAGGAAAAG-3′ for Romo1-ΔC; forward, 5′-GATCGGATCCCGCCGCCACCATGGACCGTGTCAAAATG-3′ and reverse, 5′-GATCGAATTCCTTTAGCATCGGATGCCCATCCC-3′ for Romo1-ΔN. The PCR products were cloned into pFlag-C1 vector. Dr. Xiao-Ming Yin (Beckman Research Institute, Duarte, CA, USA) kindly provided the Bcl-XL plasmid.
ROS assay
Intracellular ROS levels were measured by a fluorescence microscope (Olympus LX71 microscope; Olympus, Tokyo, Japan) or a FACScan flow cytometer (Becton Dickson, San Jose, CA, USA) using CM-H2DCFDA (50 μM) or MitoSOX (5 μM), as described earlier.26
Measurement of Δψm
To determine changes in Δψm, HeLa cells treated with TNF-α (20 ng/ml) with or without CHX (10 μg/ml) were incubated with JC-1 (5 μM) for 30 min or TMRM (100 nM) for 15 min at 37°C. JC-1 was excited at 490 nm. Emission fluorescence was filtered and images were collected for FITC (green, 530 nm) and TRITC (red, 590 nm) channels using the FACScan flow cytometer (Becton Dickson). TMRM-stained cells were examined, using a fluorescence microscope (Olympus LX71 microscope), for excitation at 510–560 nm and with an emission filter for 570–620 nm. For quantification purposes, TMRM fluorescence was analyzed with MetaMorph software (Universal Imaging, Westchester, PA, USA).
Immunoprecipitation
HEK 293 cells were plated onto 100-mm dishes and transfected with Flag-tagged Romo1. Cell lysates prepared in IP lysis buffer [50 mM Tris–HCl (pH 7.5), 120 mM NaCl, 0.5% NP-40, 50 mM NaF, 200 μM Na3VO4, 1 mM PMSF, 1 mM EDTA, 10% Glycerol] were incubated with anti-FLAG antibody (M2) or indicated antibodies. After the addition of protein G-agarose beads, cell lysates were incubated at 4°C overnight. After washing with IP lysis buffer, the immunoprecipitants were subjected to western blotting using the indicated antibodies.
Analysis of topology
Intact mitochondria were isolated from HEK 293 cells, transfected with Flag-Romo1 or Romo1-myc using an ApoAlert cell fractionation kit (Clontech, Mountain View, CA, USA), and analysis of topology was carried out as described earlier.38
Statistical analysis
Each assay was performed in triplicate and independently repeated at least three times. Statistical significance was defined as P<0.05. Means, S.E.s, and Ps were calculated using GraphPad PRISM version 4.02 for Windows (GraphPad Software, San Diego, CA, USA).
Abbreviations
- TNF-α:
-
tumor necrosis factor-α
- ROS:
-
reactive oxygen species
- Romo1:
-
ROS modulator 1
- Δψm:
-
mitochondrial membrane potential
- CHX:
-
cycloheximide
- BHA:
-
butylated hydroxy anisol
- DN-I-κB:
-
dominant negative-I-κB
- CM-H2DCFDA:
-
5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester
- JC-1:
-
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide
- TMRM:
-
tetramethylrhodamine methyl ester
References
Micheau O, Tschopp J . Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114: 181–190.
Hsu H, Xiong J, Goeddel DV . The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995; 81: 495–504.
Chen G, Goeddel DV . TNF-R1 signaling: a beautiful pathway. Science 2002; 296: 1634–1635.
Karin M, Lin A . NF-kappaB at the crossroads of life and death. Nat Immunol 2002; 3: 221–227.
Hsu H, Shu HB, Pan MG, Goeddel DV . TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 1996; 84: 299–308.
Rothe M, Wong SC, Henzel WJ, Goeddel DV . A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell 1994; 78: 681–692.
Corda S, Laplace C, Vicaut E, Duranteau J . Rapid reactive oxygen species production by mitochondria in endothelial cells exposed to tumor necrosis factor-alpha is mediated by ceramide. Am J Respir Cell Mol Biol 2001; 24: 762–768.
Meier B, Radeke HH, Selle S, Younes M, Sies H, Resch K et al. Human fibroblasts release reactive oxygen species in response to interleukin-1 or tumour necrosis factor-alpha. Biochem J 1989; 263: 539–545.
Shoji Y, Uedono Y, Ishikura H, Takeyama N, Tanaka T . DNA damage induced by tumour necrosis factor-alpha in L929 cells is mediated by mitochondrial oxygen radical formation. Immunology 1995; 84: 543–548.
Kim YS, Morgan MJ, Choksi S, Liu ZG . TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell 2007; 26: 675–687.
Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y et al. NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J 2003; 22: 3898–3909.
Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2001; 2: 222–228.
Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997; 275: 90–94.
Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M . Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005; 120: 649–661.
Gao M, Labuda T, Xia Y, Gallagher E, Fang D, Liu YC et al. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science 2004; 306: 271–275.
Muppidi JR, Tschopp J, Siegel RM . Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity 2004; 21: 461–465.
Wang L, Du F, Wang X . TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008; 133: 693–703.
Deng Y, Ren X, Yang L, Lin Y, Wu X . A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 2003; 115: 61–70.
Shirakata Y, Koike K . Hepatitis B virus X protein induces cell death by causing loss of mitochondrial membrane potential. J Biol Chem 2003; 278: 22071–22078.
Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB . Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell 1997; 91: 627–637.
Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 1995; 267: 1506–1510.
Chao DT, Linette GP, Boise LH, White LS, Thompson CB, Korsmeyer SJ . Bcl-XL and Bcl-2 repress a common pathway of cell death. J Exp Med 1995; 182: 821–828.
Gozzelino R, Sole C, Llecha N, Segura MF, Moubarak RS, Iglesias-Guimarais V et al. BCL-XL regulates TNF-alpha-mediated cell death independently of NF-kappaB, FLIP and IAPs. Cell Res 2008; 18: 1020–1036.
Gottlieb E, Vander Heiden MG, Thompson CB . Bcl-x(L) prevents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factor alpha-induced apoptosis. Mol Cell Biol 2000; 20: 5680–5689.
Chung YM, Kim JS, Yoo YD . A novel protein, Romo1, induces ROS production in the mitochondria. Biochem Biophys Res Commun 2006; 347: 649–655.
Chung YM, Lee SB, Kim HJ, Park SH, Kim JJ, Chung JS et al. Replicative senescence induced by Romo1-derived reactive oxygen species. J Biol Chem 2008; 283: 33763–33771.
Lee SB, Kim JJ, Kim TW, Kim BS, Lee MS, Yoo YD . Serum deprivation-induced reactive oxygen species production is mediated by Romo1. Apoptosis 2010; 15: 204–218.
Chung JS, Lee SB, Park SH, Kang ST, Na AR, Chang TS et al. Mitochondrial reactive oxygen species originating from Romo1 exert an important role in normal cell cycle progression by regulating p27(Kip1) expression. Free Radic Res 2009; 43: 729–737.
Na AR, Chung YM, Lee SB, Park SH, Lee MS, Yoo YD . A critical role for Romo1-derived ROS in cell proliferation. Biochem Biophys Res Commun 2008; 369: 672–678.
Hwang IT, Chung YM, Kim JJ, Chung JS, Kim BS, Kim HJ et al. Drug resistance to 5-FU linked to reactive oxygen species modulator 1. Biochem Biophys Res Commun 2007; 359: 304–310.
Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 2004; 119: 529–542.
Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ . Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 2000; 192: 1001–1014.
Kim R . Unknotting the roles of Bcl-2 and Bcl-xL in cell death. Biochem Biophys Res Commun 2005; 333: 336–343.
Lo YY, Cruz TF . Involvement of reactive oxygen species in cytokine and growth factor induction of c-fos expression in chondrocytes. J Biol Chem 1995; 270: 11727–11730.
Locksley RM, Killeen N, Lenardo MJ . The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 2001; 104: 487–501.
Matsuzawa A, Ichijo H . Stress-responsive protein kinases in redox-regulated apoptosis signaling. Antioxid Redox Signal 2005; 7: 472–481.
Jin Z, El-Deiry WS . Overview of cell death signaling pathways. Cancer Biol Ther 2005; 4: 139–163.
Cho GW, Shin SM, Kim HK, Ha SA, Kim S, Yoon JH et al. HCCR-1, a novel oncogene, encodes a mitochondrial outer membrane protein and suppresses the UVC-induced apoptosis. BMC Cell Biol 2007; 8: 50.
Acknowledgements
This work was supported by Grant FG06-2-20 from the 21st Century Frontier Functional Human Genome Project, funded by the Korean Government (MEST); a grant from the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (A084537); by the Basic Science Research Program through the National Research Foundation (NRF), funded by the Ministry of Education, Science and Technology (R01-2006-000-10113-0); and by the NRF grant funded by the MEST (SRC program, No. R11-2005-017-04001-0).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Deshmukh
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Kim, J., Lee, S., Park, J. et al. TNF-α-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-XL. Cell Death Differ 17, 1420–1434 (2010). https://doi.org/10.1038/cdd.2010.19
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2010.19
Keywords
This article is cited by
-
Rare coding variants in NOX4 link high ROS levels to psoriatic arthritis mutilans
EMBO Molecular Medicine (2024)
-
Multifaceted role of redox pattern in the tumor immune microenvironment regarding autophagy and apoptosis
Molecular Cancer (2023)
-
Holothurian triterpene glycoside cucumarioside A2-2 induces macrophages activation and polarization in cancer immunotherapy
Cancer Cell International (2023)
-
Transcriptional and functional effects of lithium in bipolar disorder iPSC-derived cortical spheroids
Molecular Psychiatry (2023)
-
Low-dose sumatriptan improves the outcome of acute mesenteric ischemia in rats via downregulating kynurenine
Pharmacological Reports (2023)
