
Abstract
SETBP1 is a SET-binding protein regulating self-renewal potential through HOXA-protein activation. Somatic SETBP1 mutations were identified by whole exome sequencing in several phenotypes of myelodysplastic/myeloproliferative neoplasms (MDS/MPN), including atypical chronic myeloid leukemia, chronic myelomonocytic leukemia, and juvenile myelomonocytic leukemia as well as in secondary acute myeloid leukemia (sAML). Surprisingly, its recurrent somatic activated mutations are located at the identical positions of germline mutations reported in congenital Schinzel–Giedion syndrome. In general, somatic SETBP1 mutations have a significant clinical impact on the outcome as poor prognostic factor, due to downstream HOXA-pathway as well as associated aggressive types of chromosomal defects (-7/del(7q) and i(17q)), which is consistent with wild-type SETBP1 activation in aggressive types of acute myeloid leukemia and leukemic evolution. Biologically, mutant SETBP1 attenuates RUNX1 and activates MYB. The studies of mouse models confirmed biological significance of SETBP1 mutations in myeloid leukemogenesis, particularly associated with ASXL1 mutations. SETBP1 is a major oncogene in myeloid neoplasms, which cooperates with various genetic events and causes distinct phenotypes of MDS/MPN and sAML.
Similar content being viewed by others


Introduction
Current advances in the development of sequencing technology enable us to identify almost all the major driver mutations in myeloid neoplasms [1,2,3,4]. In myelodysplasia including myelodysplastic syndromes (MDS) and myelodysplastic/myeloproliferative neoplasms (MDS/MPN), whole exome sequencing in about 200 cases completely revealed significant driver mutations whose frequencies were higher than 0.5% (Fig. 1) [4]. Along with these, multiple loss-of-function mutations [5,6,7,8,9,10,11,12], and recurrent activated/gain-of-function mutations [13,14,15,16] are likely to be the promising candidates for future development of novel therapy by inhibitory compounds (Fig. 2). In addition to functional significance, recent sequencing technology also uncovered clonal dynamics based on detailed information of mutated clone size. In particular, multiple samples tested at serial time points in each case can conclude acquisition timing of each mutation, clonal architecture, and intra-tumor heterogeneity in myelodysplasia [4, 17,18,19,20].

Whole exome sequencing in myelodysplasia. Mutational screening by whole exome sequencing in MDS-related myeloid neoplasms. Recurrent SETBP1 mutations are highlighted by a red rectangle. In-hose pipeline applied to mutation call is shown by sequential sky-blue flowcharts

Ideogram of major driver mutations validated by genetic and functional studies. So far, at least mutations of 54 genes were reported to be pathogenic in myelodysplasia
Using these methodologies combined with mechanistic and clinical analyses, new somatic mutations of SET-binding protein 1 (SETBP1) were discovered in MDS/MPN and secondary acute myeloid leukemia (sAML) (Fig. 3). Many follow-up studies have been conducted by various groups to clarify and detail the pathogenesis associated with SETBP1 mutations. This review article comprehensively describes genetics, biological, and clinical implication of SETBP1 mutations, including most recent research possible owing to detailed new genetic methodology and following functional investigation. Overall, of note is that recurrent SETBP1 mutations are frequently identified in distinct phenotype of myeloid neoplasms, resulting in its obvious activation of leukemogenesis.

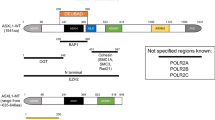
Somatic and germline mutations of SETBP1. a Somatic mutations of SETBP1 were discovered in cases with myelodysplasia. Location of the somatic mutations was identical to germline mutations of SETBP1 (asterisk), causing congenital Schinzel–Giedion syndrome (SGS). b Targeted-deep sequencing confirmed somatic status of the mutations in myeloid neoplasms
Activation of wild-type SETBP1 in myeloid neoplasms
In 2001, SETBP1 was reported as a new protein binding to SET which has an inhibitory activity for protein phosphatase 2A (PP2A) [21]. Then, SETBP1 was also found to be essential for granulocytic hematopoiesis together with EVI1 (MECOM) [22]. Before discovery of somatic mutations as below, activation of wild-type SETBP1 was already shown in various hematological neoplasms. For example, SETBP1 is a fusion partner gene in acute T cell lymphoblastic and myeloid leukemias and primary myelofibrosis (PMF) [23,24,25]. It is also a downstream target gene associated with MECOM-mediated leukemia [26]. Clinically, in cases with AML, SETBP1 overexpression was a poor prognostic factor [24]. In addition, SETBP1 locus (18q12.3) was amplified in blast phase of chronic myeloid leukemia [27]. From such multiple evidence, SETBP1 was most likely to be a major oncogene in myeloid neoplasms.
Discovery of somatic SETBP1 mutations
In December 2012, three independent studies of somatic SETBP1 mutations were presented in annual meeting of American Society of Hematology. Piazza et al. reported somatic SETBP1 mutations were frequently identified in atypical chronic myeloid leukemia (aCML) by whole exome sequencing [28]. In this study, 24% of cases with aCML were positive for mutations of this gene. Another study group from Nagoya University Department of Pediatrics (Professor Kojima group) showed that the same recurrent mutations of SETBP1 were identified in juvenile myelomonocytic leukemia (JMML) [29]. Last of the independent studies carried out in 727 cases with various myeloid malignancies by international collaboration between Cleveland Clinic and The University of Tokyo demonstrated that 7.2% of cases (n = 52) were positive for the recurrent SETBP1 mutations at Asp868, Ser869, Gly870, Ile871, and Asp880 (Fig. 3). In this large cohort, somatic SETBP1 mutations were most frequent in chronic myelomonocytic leukemia (CMML) and sAML [30]. According to no loss of heterozygosity detected at SETBP1 locus by single nucleotide polymorphism array analysis, these mutations were proved to be heterozygous. Allele-specific PCR of cDNA from mutant cases, the mRNA expression of mutated alleles was elevated compared to intact ones [30]. Since SETBP1 was supposed to be an oncogene in myeloid neoplasms, it only seems plausible to hypothesize that these SETBP1 mutations activate leukemogenic SETBP1 function. This hypothesis was proved by multiple functional studies performed using in vivo model as discussed below. Surprisingly, most of these recurrent somatic mutations are located at the same positions of germline mutations already reported in Schinzel–Giedion syndrome (Fig. 3a) [31]. This congenital disease is characterized by severe mental retardation and short-term survival [32, 33]. Since no inherited case was reported and, therefore, it is caused by de novo germline mutations [31, 34,35,36,37], this syndrome is not likely to be related to typical age-related myeloid neoplasms such as MDS/MPN and sAML. No cases with somatic SETBP1 mutations harboring symptoms of Schinzel–Giedion syndrome have been reported till date.
Disease phenotype with SETBP1 mutations
After these initial reports, many follow-up confirmatory studies of SETBP1 mutations were published in whole myeloid neoplasms. In aCML, SETBP1 mutations are most frequently identified (25–33%) [38], and in the other MDS/MPN, including JMML (8%) [29] and CMML (up to 15%) [30, 39, 40] mutations of this gene are also predominantly positive. In addition, SETBP1 mutations were also significantly more prevalent in the cases with sAML (17%) (P < 0.001) (Fig. 4a). Similarly, somatic SETBP1 mutations were secondary events following somatic or germline RAS-pathway mutations in JMML of a Japanese cohort [29], as well as in an Italian JMML cohort [41]. This also indicates that SETBP1 mutations do play a significant role rather in secondary leukemic evolution than in initial disease presentation of JMML. In analogy, secondary aCML evolution from acute myelomonocytic leukemia resulted from acquisition of SETBP1 mutation [42]. In 2 out of 25 cases with blast phase of CML, SETBP1 mutations were identified as secondary events [30]. By targeted sequencing in case series of therapy-related MDS, SETBP1 mutation was also identified [43, 44]. These findings suggest that SETBP1 mutations play a main role in secondary leukemogenesis as later subclonal events, presumably required for antecedent disease initiation by other primary genetic defects [45]. Sanger sequencing could not sufficiently detect SETBP1 mutations in CMML [39, 46] or therapy-related myeloid neoplasms [43], hence is not likely to be recommended to detect typical small clones with the mutation. In cases with another MDS/MPN subtype, refractory anemia with ring sideroblasts and thrombocytosis (RARS-T), SETBP1 mutation was also frequently observed (13%) [47].

Clinical phenotype resultant from SETBP1 mutations. High frequency of SETBP1 mutations (a) and overexpression (b) in CMML and secondary AML (non-CBF leukemia). c SETBP1 mutations are associated with loss of heterozygosity in chromosome 7/7q. d SETBP1 mutations are significantly associated with shorter overall survival
While classical myeloproliferative neoplasms (MPN), including polycythemia vera (PV), essential thrombocythemia (ET), and PMF are relatively less associated with SETBP1 mutations [48]. Another MPN subtype, chronic neutrophilic leukemia (CNL), which is closely related to CSF3R mutations [49], is also frequently affected by SETBP1 mutations (14–56%) [50,51,52]. SETBP1 mutations in CNL cases are usually subclonal events following initial CSF3R mutations with a reported exceptional case [53]. Interestingly, SETBP1 mutations are more prevalent in cases with CSF3R mutations than in those with wild-type CSF3R [50]. In fact, simultaneous decrease of allele frequencies for both CSF3R and SETBP1 mutations was found after treatment for a CNL case with Ruxolitinib [54].
Contrary to MDS/MPN or sAML, cases with de novo AML were less affected by SETBP1 mutations (frequency of mutations was <1%) (P < 0.001) (Fig. 4b). Similarly, SETBP1 mutation was not identified in childhood AML cohort [55, 56]. Overall, SETBP1 mutations are tissue-specific events acquired by myeloid lineages, and most likely to provide the cells with myeloproliferative potential.
Coordination with additional genetic events
For coordinating leukemogenesis, several genetic events were reported to be frequently detected in SETBP1-mutated cases. As described above, SETBP1 mutations are frequently mutated in CMML, aCML, and JMML, where mutations of ASXL1, SRSF2, and CBL are also prevalent (Fig. 5a). Therefore, mutations in ASXL1, SRSF2, CBL, and SETBP1 are frequently correlated with each other to present the MDS/MPN phenotypes [28, 30, 48, 57,58,59]. Among these, a synergistic effect of concomitant SETBP1 and ASXL1 mutations was functionally confirmed by the experiments using in vivo model, which revealed that ASXL1 defects initiate differentiation block and that SETBP1 activation adds proliferative potential [60]. In another context, SETBP1 mutations are also acquired by the cells which already harbor the condition of clonal expansion due to initial genetic events, for example MDS (Fig. 5b, c) [45]. Consequently, typical primary genetic events of MDS including U2AF1, and RUNX1 were also associated with SETBP1 mutations [28, 30]. However, common primary mutations of TET2 and secondary SETBP1 mutations tended to be mutually exclusive [58].

Accessory molecular events associated with SETBP1 mutations. a SETBP1 mutations coincide with various mutations of other driver genes. b Subclonal SETBP1 mutation was acquired by cells with a U2AF1- and ASXL1-mutated ancestral clone. c Serial sequencing confirmed secondary nature of SETBP1 mutations
In addition to these somatic mutations, particular chromosomal alterations are associated with SETBP1 mutations. -7/del(7q) are remarkably coincident with SETBP1 mutations, which is confirmed by multiple independent studies (Fig. 4c) [30, 45, 57, 61]. Isochromosome 17q (i(17q)) is also associated with SETBP1 mutations (54%) [45, 48, 57, 62]. Especially, TP53 and SETBP1 mutations were completely exclusive in cases with i(17q), suggesting that these two major prognostic events are independently involved in poor outcome in myeloid neoplasms [63]. Such concomitant poor prognostic chromosomal lesions are compatible with worse prognosis in cases with SETBP1 mutations as mentioned below.
Clinical implications and prognostic impacts
Clinically, SETBP1 mutations have remarkable impacts on patients’ characteristics and outcome. In a large adult cohort, SETBP1 mutations were significantly more frequent in older cases (>60 years old) (P = 0.01), cases with -7/del(7q) (P = 0.01), sAML (P < 0.001), and CMML (P = 0.002), most of which are well-known poor prognostic factors. Clustering analysis of gene-expression profiles revealed that cases with SETBP1 mutations and those with high expression of wild-type SETBP1 belonged to the closely clustered groups characterized by high expression of putatively oncogenic MECOM, TCF4, BCL11B, and DNTT genes [30], suggesting poor outcome in mutant cases. MECOM overexpression is recapitulated in a CNL case with SETBP1 mutation [64]. In fact, SETBP1 mutation is really a poor prognostic factor. In whole cohort of adult cases, overall survival is significantly shorter in cases with the mutations (HR = 2.3, 95% CI = 1.6–3.2, P < 0.001), and this tendency is more evident in younger cases (HR = 4.9, 95% CI = 2.3–5.4, P < 0.001) (Fig. 4d) [30]. These findings were reproduced in many other studies [28, 45, 57, 58, 65, 66]. By multivariable analysis including clinical factors and other genetic events as variables, SETBP1 mutation was an independent factor for overall survival (HR = 2.9, 95% CI = 1.7–4.8, P < 0.001) as well as male sex, higher age (>60 years old), and ASXL1, CBL, and DNMT3A mutations. While it was a significant worse prognostic factor by univariate analysis, -7/del(7q) abnormality was qualified after multivariable analysis because of confounding SETBP1 mutation. By following subgroup multivariable analysis in the cohort of MDS and CMML (with white blood cell count less than 12,000/μl) to which international prognostic scoring system (IPSS) score is applicable, SETBP1 mutation was an independent prognostic factor (HR = 1.8, 95% CI = 1.0–3.1, P = 0.04) as well as higher IPSS score. To further clarify a clinical effect of concomitant mutations, most significant genetic combination of CBL and SETBP1 mutations was assessed. Cases with both mutations showed significantly shorter OS than those without either of mutations [30]. Finally, in cases with JMML, secondary SETBP1 mutation was also significantly associated with poor prognosis (shorter transplantation-free survival) [29, 67]. Collectively, SETBP1 mutations are significantly associated with poor prognosis in any disease subset.
Molecular biology
Discovery of somatic SETBP1 mutations in myeloid neoplasms prompted multiple groups to clarify leukemogenic mechanisms induced by mutated SETBP1 (Fig. 6). As described above, mutant alleles are highly expressed compared to wild-type alleles [30]. In addition, protein degradation is attenuated through the mutations located at SETBP1 SKI homology region resulting in increase of protein stability [28, 30]. Therefore, at least, higher amount of mutant SETBP1 protein in cases with SETBP1 mutations are supposed to be a major consequence similar to overexpression of wild-type protein as a dose effect (Fig. 4b) [30]. Moreover, by comparison between the same amount of wild-type and mutant SETBP1 proteins, more proliferative potential was observed in the mutant experiments [30]. Mutated proteins also bind more efficiently to DNA at promotor sites of target genes [68, 69]. Altogether, SETBP1 mutations were supposed to have both quantitatively and qualitatively activating effects on SETBP1 functions.

Functional implication of activated SETBP1. Various genetic alterations were supposed to contribute to SETBP1-mediated leukemogenesis. Up- and downstream pathways, concomitant chromosomal defects, and exclusive mutations are distinctly observed in myeloid neoplasms with SETBP1 mutations
SETBP1 protein is involved in various other leukemogenic functions which were previously confirmed by various study groups. Out of these, its activated function as a transcription factor induces overexpression of HOXA9 and HOXA10, resulting in upregulation of self-renewal potential in myeloid neoplasms (Fig. 7) [30, 70]. Another SETBP1-mediated oncogenic potential is induced by down-regulation of tumor suppressor RUNX1, whose expression is attenuated through activation of SETBP1 binding more efficiently to promotor sites of RUNX1 (Fig. 8) [68]. An additional target of activated SETBP1 is a well-known oncogene MYB. Mutant forms of SETBP1 bind to MYB promotor sites and cause overexpression of this gene [69]. For these HOXA9/10, RUNX1, and MYB studies, transplantation experiments in mouse models were performed to confirm biological significance of mutated SETBP1 in myeloid leukemogenesis.

SETBP1 activation causes HOXA9/10 induction. a Setbp1 binds to Hoxa10 promoter (ChIP assay). b Hoxa10 knockdown suppresses colony formation of cells immortalized by Setbp1 transduction. c HOXA9 and HOXA10 are overexpressed in SETBP1 mutants. d HOXA9 and SETBP1 expressions correlate in patients with myeloid neoplasms

Synergism between SETBP1 and RUNX1. a Concomitant acquisition of SETBP1 and RUNX1 mutations. b RUNX1 mutations are associated with SETBP1 overexpression. c Inverse correlation between wild-type SETBP1 and RUNX1
SET-PP2A pathway is in the downstream of classical SETBP1 activation. In hematopoietic progenitor cells immortalized by forced expression of mutant SETBP1, phosphorylation of PP2A was accelerated (Fig. 6) [28, 30]. Mouse transplantation model of mutated SETBP1 also showed PP2A inhibition and HOXA9/10 activation [60]. These findings suggest that phosphorylated (loss-of-function) PP2A could be a therapeutic target of PP2A activators.
A most recent study has proposed very surprising but interesting concepts by comparing somatic and germline SETBP1 mutations [71]. They described that consequence of SETBP1 mutations are different among substitutions in SETBP1 residues. Mutations at I871 resulted in a weak increase in protein levels and are significantly more frequent in Schinzel–Giedion syndrome (germline) than in myeloid neoplasms (somatic). On the other hand, substitutions in residue D868 led to the largest increase in protein levels. Cases with germline mutations affecting D868 have enhanced cell proliferation in vitro and higher incidence of cancer compared to patients with other germline SETBP1 mutations [71]. While these updated functional studies are helpful to understand biological mechanisms of leukemogenesis, therapeutic strategy to this distinct molecular target, SETBP1 mutation, is not established yet and requires further investigation.
Summary
Approximately 13 years after cloning SETBP1, this gene is now commonly recognized as a driver oncogene almost exclusively in myeloid neoplasms. Somatic mutations in myeloid neoplasms are identical to germline mutations in Schinzel–Giedion syndrome. Activating SETBP1 mutations is a poor prognostic factor and frequently acquired as secondary event. Functionally, these mutations are involved in various leukemogenic mechanisms through phosphorylated PP2A, HOXA clusters, RUNX1, MYB, other associated mutations, and chromosomal abnormalities. Activation of SETBP1 results in a distinct disease entity and inhibition of this protein as a molecular target should be an attractive therapeutic strategy.
Change history
23 October 2021
A Correction to this paper has been published: https://doi.org/10.1007/s12185-021-03236-1
References
Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379–90.
Makishima H, Yoshizato T, Yoshida K, Sekeres MA, Radivoyevitch T, Suzuki H, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet. 2017;49:204–12.
Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–301.
Makishima H, Jankowska AM, Tiu RV, Szpurka H, Sugimoto Y, Hu Z, et al. Novel homo- and hemizygous mutations in EZH2 in myeloid malignancies. Leukemia. 2010;24:1799–804.
Grossmann V, Tiacci E, Holmes AB, Kohlmann A, Martelli MP, Kern W, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118:6153–63.
Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet. 2013;45:1232–7.
Hosono N, Makishima H, Jerez A, Yoshida K, Przychodzen B, McMahon S, et al. Recurrent genetic defects on chromosome 7q in myeloid neoplasms. Leukemia. 2014;28:1348–51.
Kurtovic-Kozaric A, Przychodzen B, Singh J, Konarska MM, Clemente MJ, Otrock ZK, et al. PRPF8 defects cause missplicing in myeloid malignancies. Leukemia. 2015;29:126–36.
Huang D, Nagata Y, Grossmann V, Radivoyevitch T, Okuno Y, Nagae G, et al. BRCC3 mutations in myeloid neoplasms. Haematologica. 2015;100:1051–7.
Mori T, Nagata Y, Makishima H, Sanada M, Shiozawa Y, Kon A, et al. Somatic PHF6 mutations in 1760 cases with various myeloid neoplasms. Leukemia. 2016;30:2270–3.
Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009;460:904–8.
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9.
Gomez-Segui I, Makishima H, Jerez A, Yoshida K, Przychodzen B, Miyano S, et al. Novel recurrent mutations in the RAS-like GTP-binding gene RIT1 in myeloid malignancies. Leukemia. 2013;27:1943–6.
Yoda A, Adelmant G, Tamburini J, Chapuy B, Shindoh N, Yoda Y, et al. Mutations in G protein beta subunits promote transformation and kinase inhibitor resistance. Nat Med. 2015;21:71–5.
Meggendorfer M, de Albuquerque A, Nadarajah N, Alpermann T, Kern W, Steuer K, et al. Karyotype evolution and acquisition of FLT3 or RAS pathway alterations drive progression of myelodysplastic syndrome to acute myeloid leukemia. Haematologica. 2015;100:e487–90.
Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366:1090–8.
Kim T, Tyndel MS, Kim HJ, Ahn JS, Choi SH, Park HJ, et al. The clonal origins of leukemic progression of myelodysplasia. Leukemia. 2017; doi:10.1038/leu.2017.17.
Pellagatti A, Roy S, Di Genua C, Burns A, McGraw K, Valletta S, et al. Targeted resequencing analysis of 31 genes commonly mutated in myeloid disorders in serial samples from myelodysplastic syndrome patients showing disease progression. Leukemia. 2016;30:247–50.
Minakuchi M, Kakazu N, Gorrin-Rivas MJ, Abe T, Copeland TD, Ueda K, et al. Identification and characterization of SEB, a novel protein that binds to the acute undifferentiated leukemia-associated protein SET. Eur J Biochem. 2001;268:1340–51.
Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–9.
Panagopoulos I, Kerndrup G, Carlsen N, Strombeck B, Isaksson M, Johansson B. Fusion of NUP98 and the SET binding protein 1 (SETBP1) gene in a paediatric acute T cell lymphoblastic leukaemia with t(11;18)(p15;q12). Br J Haematol. 2007;136:294–6.
Cristobal I, Blanco FJ, Garcia-Orti L, Marcotegui N, Vicente C, Rifon J, et al. SETBP1 overexpression is a novel leukemogenic mechanism that predicts adverse outcome in elderly patients with acute myeloid leukemia. Blood. 2010;115:615–25.
Albano F, Anelli L, Zagaria A, Coccaro N, Casieri P, Minervini A, et al. SETBP1 and miR_4319 dysregulation in primary myelofibrosis progression to acute myeloid leukemia. J Hematol Oncol. 2012;5:48.
Goyama S, Yamamoto G, Shimabe M, Sato T, Ichikawa M, Ogawa S, et al. Evi-1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell. 2008;3:207–20.
Makishima H, Jankowska AM, McDevitt MA, O’Keefe C, Dujardin S, Cazzolli H, et al. CBL, CBLB, TET2, ASXL1, and IDH1/2 mutations and additional chromosomal aberrations constitute molecular events in chronic myelogenous leukemia. Blood. 2011;117:e198–206.
Piazza R, Valletta S, Winkelmann N, Redaelli S, Spinelli R, Pirola A, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet. 2013;45:18–24.
Sakaguchi H, Okuno Y, Muramatsu H, Yoshida K, Shiraishi Y, Takahashi M, et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet. 2013;45:937–41.
Makishima H, Yoshida K, Nguyen N, Przychodzen B, Sanada M, Okuno Y, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45:942–6.
Hoischen A, van Bon BW, Gilissen C, Arts P, van Lier B, Steehouwer M, et al. De novo mutations of SETBP1 cause Schinzel–Giedion syndrome. Nat Genet. 2010;42:483–5.
Schinzel A, Giedion A. A syndrome of severe midface retraction, multiple skull anomalies, clubfeet, and cardiac and renal malformations in sibs. Am J Med Genet. 1978;1:361–75.
Herenger Y, Stoetzel C, Schaefer E, Scheidecker S, Maniere MC, Pelletier V, et al. Long term follow up of two independent patients with Schinzel–Giedion carrying SETBP1 mutations. Eur J Med Genet. 2015;58:479–87.
Carvalho E, Honjo R, Magalhaes M, Yamamoto G, Rocha K, Naslavsky M, et al. Schinzel–Giedion syndrome in two Brazilian patients: report of a novel mutation in SETBP1 and literature review of the clinical features. Am J Med Genet A. 2015;167A:1039–46.
Ko JM, Lim BC, Kim KJ, Hwang YS, Ryu HW, Lee JH, et al. Distinct neurological features in a patient with Schinzel–Giedion syndrome caused by a recurrent SETBP1 mutation. Childs Nerv Syst. 2013;29:525–9.
Suphapeetiporn K, Srichomthong C, Shotelersuk V. SETBP1 mutations in two Thai patients with Schinzel–Giedion syndrome. Clin Genet. 2011;79:391–3.
Takeuchi A, Okamoto N, Fujinaga S, Morita H, Shimizu J, Akiyama T, et al. Progressive brain atrophy in Schinzel–Giedion syndrome with a SETBP1 mutation. Eur J Med Genet. 2015;58:369–71.
Gotlib J. How I treat atypical chronic myeloid leukemia. Blood. 2017;129:838–45.
Laborde RR, Patnaik MM, Lasho TL, Finke CM, Hanson CA, Knudson RA, et al. SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia (CMML): independent prognostic impact in CMML. Leukemia. 2013;27:2100–2.
Ouyang Y, Qiao C, Chen Y, Zhang SJ. Clinical significance of CSF3R, SRSF2 and SETBP1 mutations in chronic neutrophilic leukemia and chronic myelomonocytic leukemia. Oncotarget. 2017. doi:10.18632/oncotarget.5355.
Bresolin S, De Filippi P, Vendemini F, D’Alia M, Zecca M, Meyer LH, et al. Mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia: a report from the Italian AIEOP study group. Oncotarget. 2016;7:28914–9.
Hu W, Wang X, Yang R, Xie Y, Zhang Z, Lu H, et al. A novel mutation of SETBP1 in atypical chronic myeloid leukemia transformed from acute myelomonocytic leukemia. Clin Case Rep. 2015;3:448–52.
Fabiani E, Falconi G, Fianchi L, Criscuolo M, Leone G, Voso MT. SETBP1 mutations in 106 patients with therapy-related myeloid neoplasms. Haematologica. 2014;99:e152–3.
Fabiani E, Falconi G, Fianchi L, Criscuolo M, Ottone T, Cicconi L, et al. Clonal evolution in therapy-related neoplasms. Oncotarget. 2017. doi:10.18632/oncotarget.4509.
Hou HA, Kuo YY, Tang JL, Chou WC, Yao M, Lai YJ, et al. Clinical implications of the SETBP1 mutation in patients with primary myelodysplastic syndrome and its stability during disease progression. Am J Hematol. 2014;89:181–6.
Cui Y, Tong H, Du X, Li B, Gale RP, Qin T, et al. Impact of TET2, SRSF2, ASXL1 and SETBP1 mutations on survival of patients with chronic myelomonocytic leukemia. Exp Hematol Oncol. 2015;4:14.
Patnaik MM, Lasho TL, Finke CM, Hanson CA, King RL, Ketterling RP, et al. Predictors of survival in refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) and the role of next-generation sequencing. Am J Hematol. 2016;91:492–8.
Meggendorfer M, Bacher U, Alpermann T, Haferlach C, Kern W, Gambacorti-Passerini C, et al. SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia. 2013;27:1852–60.
Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368:1781–90.
Gotlib J, Maxson JE, George TI, Tyner JW. The new genetics of chronic neutrophilic leukemia and atypical CML: implications for diagnosis and treatment. Blood. 2013;122:1707–11.
Pardanani A, Lasho TL, Laborde RR, Elliott M, Hanson CA, Knudson RA, et al. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia. 2013;27:1870–3.
Cui Y, Li B, Gale RP, Jiang Q, Xu Z, Qin T, et al. CSF3R, SETBP1 and CALR mutations in chronic neutrophilic leukemia. J Hematol Oncol. 2014;7:77.
Lasho TL, Mims A, Elliott MA, Finke C, Pardanani A, Tefferi A. Chronic neutrophilic leukemia with concurrent CSF3R and SETBP1 mutations: single colony clonality studies, in vitro sensitivity to JAK inhibitors and lack of treatment response to ruxolitinib. Leukemia. 2014;28:1363–5.
Nooruddin Z, Miltgen N, Wei Q, Schowinsky J, Pan Z, Tobin J, et al. Changes in allele frequencies of CSF3R and SETBP1 mutations and evidence of clonal evolution in a chronic neutrophilic leukemia patient treated with ruxolitinib. Haematologica. 2017. doi:10.3324/haematol.2016.163089.
Choi HW, Kim HR, Baek HJ, Kook H, Cho D, Shin JH, et al. Alteration of the SETBP1 gene and splicing pathway genes SF3B1, U2AF1, and SRSF2 in childhood acute myeloid leukemia. Ann Lab Med. 2015;35:118–22.
Shiba N, Ohki K, Park MJ, Sotomatsu M, Kudo K, Ito E, et al. SETBP1 mutations in juvenile myelomonocytic leukaemia and myelodysplastic syndrome but not in paediatric acute myeloid leukaemia. Br J Haematol. 2014;164:156–9.
Fernandez-Mercado M, Pellagatti A, Di Genua C, Larrayoz MJ, Winkelmann N, Aranaz P, et al. Mutations in SETBP1 are recurrent in myelodysplastic syndromes and often coexist with cytogenetic markers associated with disease progression. Br J Haematol. 2013;163:235–9.
Damm F, Itzykson R, Kosmider O, Droin N, Renneville A, Chesnais V, et al. SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias. Leukemia. 2013;27:1401–3.
Patnaik MM, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA, et al. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia. 2014;28:2206–12.
Inoue D, Kitaura J, Matsui H, Hou HA, Chou WC, Nagamachi A, et al. SETBP1 mutations drive leukemic transformation in ASXL1-Mutated MDS. Leukemia. 2015;29:847–57.
Xu F, Wu LY, He Q, Wu D, Zhang Z, Song LX, et al. Exploration of the role of gene mutations in myelodysplastic syndromes through a sequencing design involving a small number of target genes. Sci Rep. 2017;7:43113.
Kanagal-Shamanna R, Luthra R, Yin CC, Patel KP, Takahashi K, Lu X, et al. Myeloid neoplasms with isolated isochromosome 17q demonstrate a high frequency of mutations in SETBP1, SRSF2, ASXL1 and NRAS. Oncotarget. 2016;7:14251–8.
Adema V, Larrayoz MJ, Calasanz MJ, Palomo L, Patino-Garcia A, Agirre X, et al. Correlation of myelodysplastic syndromes with i(17)(q10) and TP53 and SETBP1 mutations. Br J Haematol. 2015;171:137–41.
Altangerel O, Cao S, Meng J, Liu P, Haiyan G, Xu Y, et al. Chronic neutrophilic leukemia with overexpression of EVI-1, and concurrent CSF3R and SETBP1 mutations: a case report. Oncol Lett. 2015;10:1694–700.
Thol F, Josef Suchanek K, Koenecke C, Stadler M, Platzbecker U, Thiede C, et al. SETBP1 mutation analysis in 944 patients with MDS and AML. Leukemia. 2013;27:2072–5.
Shou LH, Cao D, Dong XH, Fang Q, Wu Y, Zhang Y, et al. Prognostic significance of SETBP1 mutations in myelodysplastic syndromes, chronic myelomonocytic leukemia, and chronic neutrophilic leukemia: a meta-analysis. PLoS One. 2017;12:e0171608.
Stieglitz E, Troup CB, Gelston LC, Haliburton J, Chow ED, Yu KB, et al. Subclonal mutations in SETBP1 confer a poor prognosis in juvenile myelomonocytic leukemia. Blood. 2015;125:516–24.
Vishwakarma BA, Nguyen N, Makishima H, Hosono N, Gudmundsson KO, Negi V, et al. Runx1 repression by histone deacetylation is critical for Setbp1-induced mouse myeloid leukemia development. Leukemia. 2016;30:200–8.
Nguyen N, Vishwakarma BA, Oakley K, Han Y, Przychodzen B, Maciejewski JP, et al. Myb expression is critical for myeloid leukemia development induced by Setbp1 activation. Oncotarget. 2016;7:86300–12.
Oakley K, Han Y, Vishwakarma BA, Chu S, Bhatia R, Gudmundsson KO, et al. Setbp1 promotes the self-renewal of murine myeloid progenitors via activation of Hoxa9 and Hoxa10. Blood. 2012;119:6099–108.
Acuna-Hidalgo R, Deriziotis P, Steehouwer M, Gilissen C, Graham SA, van Dam S, et al. Overlapping SETBP1 gain-of-function mutations in Schinzel–Giedion syndrome and hematologic malignancies. PLoS Genet. 2017;13:e1006683.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest to disclose.
About this article

Cite this article
Makishima, H. Somatic SETBP1 mutations in myeloid neoplasms. Int J Hematol 105, 732–742 (2017). https://doi.org/10.1007/s12185-017-2241-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2241-1