
Abstract
Wernicke’s Encephalopathy is an acute neuro-psychiatric condition caused by an insufficient supply of thiamine (Vitamin B1) to the brain. If undiagnosed or inadequately treated, it is likely to proceed to Korsakoff’s Syndrome. Wernicke’s Encephalopathy can result from dietary deficiency alone and this form is usually successfully treated, with little chance of Korsakoff’s Syndrome supervening. On the other hand, thiamine deficiency associated with alcohol misuse/dependence may require up to 1 gram of thiamine IV in the first 24 hours to be treated successfully. The reasons for this difference in treatment will be discussed. Thiamine diphosphate acts as a co-factor for a number of thiamine-dependent enzymes. Thiamine deficiency leads to a reduction in the activity of these enzymes, and this leads to alterations in mitochondrial activity, impairment of oxidative metabolism, decreased energy status and eventually selective neuronal death. The damage caused by the combination of thiamine deficiency and alcohol metabolism probably interferes with adequate thiamine transport at a number of sites in the body, including the blood–brain barrier, as well as causing damage to the apoenzymes which then require higher concentrations of thiamine to work normally. The accumulated damage is likely to render the use of oral thiamine therapeutically inadequate since the body is unable to produce high enough concentrations of thiamine in the blood to traverse the blood–brain barrier. Some individuals are probably genetically predisposed to develop Wernicke’s. Long before individuals with alcohol misuse or dependence develop Wernicke’s Encephalopathy the neurons and other cells of the body are functioning sub-optimally because of the inadequate supply of thiamine and the neurotoxic effect of alcohol. This relative deficiency initiates a series of pathological changes which accumulate and further interfere with the supply of thiamine and its utilisation at a time when the requirements are increased. The best treatment for Korsakoff’s Syndrome is timely recognition of Wernicke’s Encephalopathy and appropriate intervention and prevention.
Similar content being viewed by others


Introduction
Korsakoff’s syndrome (KS) has been defined by Victor et al. (1971) as “an abnormal mental state in which memory and learning are affected out of all proportion to other cognitive functions in an otherwise alert and responsive patient”. Adaptation to every new situation requires the acquisition of new information and its integration with past experience. Failure to make new memories renders the patient capable of performing only the most habitual routines. KS usually follows Wernicke’s encephalopathy (WE) although evidence of this preceding event is not always documented. The challenge is to prevent the patient developing KS but if this fails, to manage KS so as to improve brain function and to aid the patient to adapt to their cognitive impairment.
The Natural History of Korsakoff’s Syndrome: Wernicke’s Encephalopathy
Wernicke’s encephalopathy (WE) is an acute neuro-psychiatric condition caused by an inadequate supply of thiamine (vitamin B1) to the brain. It frequently but not invariably occurs in individuals with alcohol problems. Thiamine deficiency can occur as a result of inadequate dietary intake, reduced absorption of thiamine, and a reduction in the rate of conversion to the active metabolite. One or more of these factors may be implicated. WE is reversible if treated with a timely and adequate dose of parenteral thiamine. If it is undiagnosed or inadequately treated, it is likely to proceed to the chronic state, Korsakoff’s Syndrome (KS). It follows from this that the best treatment for KS is timely recognition of WE, and appropriate intervention and prevention.
When WE is the result of thiamine deficiency alone, e.g., in prisoners of war, in hyperemesis gravidarum or gastric cancer, treatment with small doses of oral/subcutaneous thiamine is usually successful, and very few individuals progress to KS. When WE occurs in individuals with an alcohol problem the situation is more complicated, because alcohol is thought to compromise thiamine transport systems and to interfere with thiamine utilization. These changes mean that higher doses of thiamine are required to treat the condition successfully in alcohol misusers, and the thiamine must be given parenterally. Unfortunately KS is a frequent outcome in these patients and the reasons for this will be discussed.
Alcohol Related Brain Damage
Long term heavy alcohol consumption is known to lead to cognitive impairment and these effects are variously described in the literature as “alcohol-related dementia”; “alcoholic dementia”; “alcohol-induced dementia”; alcohol-related cognitive impairment”; “alcohol-related brain damage” (ARBD). Individuals with alcohol misuse and Wernicke-Korsakoff Syndrome (WKS) typically show evidence of cerebral damage as a result both of thiamine deficiency and alcohol-induced neurotoxicity. In addition to this, the synergistic effects of thiamine deficiency and alcohol-induced neurotoxicity can also have profound and lasting effects. It therefore follows that WKS may be a more heterogeneous condition than has been previously appreciated, and affected individuals, who present with a spectrum of brain damage, have a range of therapeutic requirements (Zahr et al. 2011).
It has been estimated that about 10 % of the dementia population has an “alcohol- related dementia” and that this figure is likely to be higher in areas of socio-economic deprivation (Lishman 1990; MacRae and Cox 2003). It is difficult to estimate accurately the prevalence of ARBD because this is a “hidden” and marginalised population of uncertain size, frequently undiagnosed and living in nursing homes and hostels, and without financial resources or access to specialist evaluation. In recent years there has been some evidence of increased rates of ARBD in areas of heavy alcohol use and social deprivation, for instance in Glasgow, Scotland (Ramayya and Jauhar 1997). A survey of a homeless hostel population in Glasgow (n = 4000) further estimated that 21 % of “residents” showed evidence of some degree of ARBD (Gilchrist and Morrison 2005). In Scotland most individuals with ARBD who are known to treatment services are in their 50s or early 60s (MacRae and Cox 2003). Ages are lower in Australia where there is greater emphasis on early identification (Price et al. 1988). Because the ARBD population is younger than the typical dementia population, it has different needs and this has resource implications for management and rehabilitation. The general hospital setting is often the first point of contact for such individuals, so staff in the Emergency Room should be alert to the range of presentations, which may be legion.
Autopsy studies in general hospital populations have shown that 1.5 % of all patients specifically examined post-mortem for damage in the WKS areas have neuropathological brain lesions characteristic of the syndrome and that 12.5 % of alcoholics have these brain lesions (Harper 2006; Harper et al. 1988, 2003). This figure increases to 35 % when thiamine- dependent cerebellar damage is included (Torvik et al. 1982).
A recent survey indicated that almost 1 in 3 patients being admitted to hospitals in the UK showed evidence of ‘malnutrition’, and that much of this malnutrition originated in the community (Russell and Elia 2009). Although common across all age groups and diagnostic categories, malnutrition was more prevalent in women (who were older) and in patients over 65 years. Certain diagnostic categories were over-represented, including gastrointestinal and neurological disease.
The prevention of KS and ARBD is therefore a significant public health issue, both in developed and developing countries. This is not only a matter of promoting a healthy diet. A greater understanding of the effects of alcohol consumption on public health needs to be appreciated by governments responsible for setting the policy agenda. It is important that attitudes to alcohol, alcohol problems and social deprivation are also tackled.
Intoxication, Withdrawal and Thiamine Deficiency in the “Alcoholic” Brain
Alcohol is a non-specific drug which interacts with a number of neurotramsmitter pathways, including the gamma-aminobutyric acid (GABA) and glutamate systems, which underpin the development of tolerance and dependence (Marshall et al. 2010). GABA is the major inhibitory system in the brain and glutamate the major excitatory system. Alcohol enhances the inhibitory GABA receptors and inhibits the excitatory glutamate receptors (the N-methyl-D-aspartic acid (NMDA) subtype), and chronic heavy alcohol consumption leads to neuroadaptation in these systems, with a compensatory reduction in GABA function and an increase in glutamate function. When an alcohol dependent individual suddenly stops drinking, this system is thrown into imbalance and signs and symptoms of alcohol withdrawal emerge. In extreme cases of withdrawal, patients develop alcohol withdrawal seizures (AWS) and delirium tremens (DT). Cortisol levels are elevated during alcohol withdrawal, signifying that it is a stressful process (Mendelson and Stein 1966; Keedwell et al. 2001).
Repeated bouts of drinking and withdrawal are likely to lead to glutamate-induced excitability and lasting neuronal damage (Brust 2010). The demand for thiamine is likely to be increased in the alcoholic brain because of increased requirements for alcohol metabolism, which may be further increased during alcohol withdrawal. Thiamine deficiency also causes excessive glutamate release so when it occurs in combination with alcohol- induced glutamate receptor upregulation, the potential for neurotoxicity is increased. It is no accident that case reports describing episodes of WE show that it typically occurs during a period of sudden unexpected and untreated alcohol withdrawal.
If thiamine deficiency supervenes during alcohol withdrawal, it is unlikely to be immediately reversed by the administration of oral or parenteral thiamine alone. Because of other nutritional deficiencies and cellular disruption, ongoing abstinence from alcohol and good nutrition are also required (Thomson 2000; Thomson et al. 2002). It is not known how long this state persists, and it is likely to vary from one patient to another. A normal circulating concentration of thiamine diphosphate (TDP) in the blood does not guarantee that adequate amounts of thiamine are reaching the enzymes of the brain cells. Further work is needed to optimise the treatment of WE and to prevent KS.
Thiamine Deficiency in Animals With and Without Alcohol: Implications for Humans
There are two types of experimental animal model of thiamine deficiency (TD): drug-induced TD caused by an antithiamine drug such as pyrithiamine or oxythiamine, and dietary-induced TD. Experiments in animals do not exactly replicate what happens in humans, but they give an insight into what happens in pure thiamine deficiency, with and without alcohol. Dietary thiamine deficiency in rhesus monkeys was associated with the development of lesions in some structures (inferior colliculus and medical vestibular nuclei) after only one period of thiamine deprivation whereas other structures were more resistant (Witt and Goldman-Rakic 1983a). Damage to the basal ganglia was associated with lengthy episodes rather than with frequent periods of deprivation. The mammillary bodies and dorsomedial nucleus of the thalamus were spared suggesting that lesions in these structures occur only in the most advanced stages of thiamine deprivation (Witt and Goldman-Rakic 1983a). Although no neuronal degeneration was seen in the mammillary bodies and dorsomedial nucleus of the thalamus in these monkeys, they showed evidence of memory deficits similar to those seen in WKS patients (Witt and Goldman-Rakic 1983b).
Damage due to alcohol appears to be more severe in the presence of thiamine deficiency and recurrent periods of thiamine deficiency cause cumulative brain damage (Crowe and El-Hadj 2002; Price et al. 1993; Ciccia and Langlais 2000).
The implications of these findings are that alcohol dependent patients should be given prophylactic thiamine as a matter of course, orally or parenterally (if absorption is impaired) to help protect the brain during episodes of detoxification or medically assisted withdrawal or whenever they present to medical services. Their ability to absorb thiamine orally can be checked by demonstrating an adequate increase in the circulating thiamine diphosphate (TDP), although this is not available as a routine or rapid test. If this is insufficient and they remain at risk (Thomson et al. 2002; Thomson and Marshall 2006) it is recommended that they receive 250 mg of thiamine hydrochloride (Pabrinex in UK) IM daily for 5 days. This dose of thiamine has not been evaluated by controlled trials and there is currently no established threshold level of TDP above which all patients would be safe from developing brain damage (Thomson et al. 2009).
While animal models can deal with specific and possible mechanisms they cannot reflect the complexity of the interaction of thiamine and other vitamin deficiencies in individuals with varying degrees of alcohol dependence, intoxication and withdrawal. This is not easily replicable in animals.
Korsakoff’s Syndrome (KS) is unlikely to be caused purely by thiamine deficiency. Failure to respond to thiamine may in part be due to the fact that cells have been irreversibly damaged by lack of thiamine, but could also be due to deficiencies of other nutrients and alcohol-induced neurotoxicity. There have been no trials of other nutrients and indeed, no studies have been done to test whether WE, treated with the full recommended dose of thiamine, prevents the development of KS or reduces its incidence.
Factors Affecting the Availability of Thiamine to the Brain in Alcohol Dependence (Table 1)
Genetic Predisposition
Some individuals are probably genetically predisposed to developing WE. Alcoholics may have genetic defects in thiamine transport which only become evident if they drink sufficient alcohol thereby limiting the availability of thiamine and causing problems with transport systems and storage. Two genes involved in the cellular transport of thiamine have recently been established: THTR1 (product SLC19A2) the high affinity transporter and THTR2 (product SLCA3) the low affinity transporter and we have identified changes in the genes of some WE patients which could potentially affect gene expression (Guerrini et al. 2005, 2009).
Subramanian et al. (2010) have reported that chronic alcohol consumption in rats fed a Lieber-DeCarli diet (a nutritionally adequate liquid diet containing ethanol (Lieber and De Carli 1989)) was associated with decreased carrier-mediated thiamine transport across the renal brush-border and basolateral membranes and in transcriptionally-mediated inhibition of the THTR1 and THTR2 expression. In addition, the expression of thiamin pyrophosphokinase (TPKase), the rate-limiting enzyme in the synthesis of the coenzyme form of thiamine, was significantly reduced. They also found that chronic alcohol administration reduced intestinal thiamine absorption in the rat which was accompanied by a decrease in transcription and expression of the Slc19a2 and Slc19a3 genes (Subramanya et al. 2010) confirming the observations made in man in the 1960s (Thomson 1966; Thomson et al. 1970; Thomson and Leevy 1972). This will be discussed further in the section on genetic susceptibility.
Inadequate Thiamine Intake
Individuals with alcohol dependence typically neglect their diet and eat poorly. Often ingested nutrients are lost through vomiting, diarrhoea or steatorrhoea. The total body stores of thiamine are small, being approximately 30 mg (daily requirements 1–2 mg, increased by alcohol metabolism) and will be depleted within a few weeks by poor intake even if absorption from the gastrointestinal tract remains normal.
Impaired Absorption of Thiamine
It was demonstrated in the 1960s that alcohol can interfere with the absorption of thiamine in man which requires an active transport system. Further malabsorption occurs as malnutrition becomes a significant additional factor (Thomson 1966; Thomson et al. 1968, 1970; Thomson and Leevy 1972). Alcohol-related liver damage reduces storage of thiamine, and thiamine phosphorylation is impaired. When malabsorption due to alcohol and malnutrition becomes a significant factor, this accelerates the decline, often rendering treatment with oral thiamine inadequate (Thomson 2000; Thomson and Marshall 2006).
Thiamine Transport Problems
The damage caused by the combination of thiamine deficiency and alcohol metabolism probably interferes with adequate thiamine transport at a number of sites in the body, including the blood–brain barrier, as well as causing damage to the apoenzymes which then require higher concentrations of thiamine to work normally (Pratt et al. 1985: Heap et al. 2002). The accumulated damage means that treatment with oral thiamine is inadequate since it is unable to produce high enough concentrations of thiamine in the blood to traverse the blood–brain barrier (Thomson et al. 2002). This is in marked contrast to the findings in WE due to dietary deficiency alone, as these individuals do not seem to have significant problems with thiamine transport and therefore respond to small doses of thiamine.
Other Nutritional Deficiencies
In addition to the effects of ethanol on the intestinal transport of thiamine, other nutritional deficiencies found in chronic alcoholic patients may also impair absorption. Folate deficiency is associated with a decreased thiamine content in blood and liver in rats, suggesting concomitant impaired absorption of thiamine. Thomson et al. (1972) found that folate deficient rats absorbed thiamine less efficiently and the data indicated that folate played a role in maintaining the integrity of the active transport process (Howard et al. 1974). In rats, deficiencies of Vitamins B6 and B12 can lead to decreased stores of thiamine (Nishino and Itokawa 1977) and thiamine itself may alter the kinetics of thiamine transport (Hoyumpa et al. 1976).
These findings emphasise the importance of replacing all deficient nutrients when treating these patients. Limited studies in humans indicate that at physiological levels of thiamine intake and even with oral thiamine supplementation, absorption is dependent on the active transport system which may be significantly impaired by the factors mentioned above (Thomson and Leevy 1972). Unlike the absorption of thiamine propyl disulfide which occurs by diffusion in man, this has not been demonstrated for the water soluble compounds like thiamine hydrochloride (Thomson et al. 1971, 1983; Baker et al. 1974).
It is possible that this mechanism may also interfere with transport of thiamine into the neurone. Because folate stimulates neurogenesis and because this has been shown to be impaired in the hippocampus in the pre-pathological lesion stage of WKS, impaired folate transport could also be a mechanism for cognitive damage. Similarly hepatic encephalopathy and hypoglycaemia could also be contributory factors. Acker et al. (1982) demonstrated that in chronic alcoholics topographical brain changes became more pronounced with increasing severity of liver injury. Successful prevention and treatment of KS will depend upon replacing all nutrients that are deficient and attempting to prevent or repair damage due to neurotoxicity. Recent proteomics studies showed that, in alcoholics with cirrhosis of the liver, the brain expression of the thiamine-related enzyme transketolase is significantly reduced when compared to controls or to alcoholics without cirrhosis of the liver (Matsumoto 2009).
Effects of Low Circulating Thiamine Levels
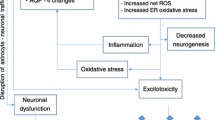
Numerous studies of alcoholic patient groups in different countries have demonstrated that many individuals have low circulating blood thiamine levels (Leevy et al. 1965; Thomson et al. 1987; Cook et al. 1998; Mancinelli et al. 2003; Brust 2010). Thiamine diphosphate (TDP) acts as a co-factor for a number of thiamine-dependent enzymes. Thiamine deficiency leads to a reduction in the activity of these enzymes, and this further leads to alterations in mitochondrial activity, impairment of oxidative metabolism, decreased energy status and eventually selective neuronal death.
The Contribution of Alcoholic Neurotoxicity to the Development of KS
A number of authors have suggested that thiamine deficiency may interact synergistically with alcohol neurotoxicity to promote progression from WE to Korsakoff’s Syndrome (Wood 1985; Victor and Adams 1985; Victor et al. 1989). Crowe and Kempton (1997) demonstrated that the combination of thiamine deficiency (induced by oxythiamine) and alcohol neurotoxicity in young chicks interacted to impair memory retention. This impairment was irreversible and did not respond to treatment with thiamine, unlike the deficits observed in chicks who were only subjected to thiamine depletion. In a later experiment, phenytoin was given immediately after the training experiment and was shown to improve memory deficits induced in the chicks by ethanol toxicity in combination with thiamine deficiency (Crowe and El-Hadj 2002). Given that phenytoin is a facilitator of NA+/K + ATPase activity it was postulated that the combined effect of oxythiamine and ethanol exposure was interference with Na/K ATPase activity.
Genetics
The importance of thiamine to the central nervous system is shown by its key role in glucose and energy metabolism. Eighty percent of brain thiamine is in the form of thiamine diphosphate, a cofactor for three thiamine dependent enzymes important in brain cell metabolism – the α ketoglutarate dehydrogenase complex, transketolase and pyruvate dehydrogenase enzymes. Butterworth and colleagues demonstrated significant reductions of the thiamine-dependent enzymes: pyruvate dehydrogenase complex, alpha-ketoglutarate dehydrogenase and transketolase, in autopsied cerebellar vermis samples from alcoholic patients with the clinically and neuropathologically confirmed diagnosis of Wernicke-Korsakoff syndrome (Butterworth et al. 1993).
In the scientific literature it has been repeatedly hypothesized that there is a heritable susceptibility to thiamine deficiency comparable to other hereditary metabolic disorders, and a genetic component in the pathogenesis of Wernicke Korsakoff Syndrome has been postulated since the late 1970s. Since then very few genetic studies have been carried out on candidate genes such as thiamine dependent enzymes, alcohol metabolizing enzymes and GABA receptors. The findings are controversial and not conclusive (Guerrini et al. 2009).
Several authors reported the important role of the thiamine transporters in the pathogenesis of the thiamine deficiency disorders. In humans, thiamine cannot be synthesized and must therefore be obtained from exogenous sources, via absorption in the intestine. The cellular transport of thiamine is mediated by specific carriers, recently characterised: the Thiamine Transporter-1 and Transporter-2, respectively the products of the SLC19A2 and SLC19A3 genes (Guerrini et al. 2009). Gene expression studies have shown that the thiamine transporters are well expressed in several tissues such the intestine, placenta, kidneys and brain (Guerrini et al. 2005).
The SLC19A2 gene encodes the human thiamine transporter protein. It is made up of 497 amino acids and is predicted to have 12 transmembrane domains responsible for importing thiamine into the cell for utilisation by thiamine dependent enzymes such as 2-oxoglutarate dehydrogenase complex. Recently, Diaz and colleagues (1999) mapped the high affinity thiamine transporter gene to chromosome 1q23.2-q23.3 (Guerrini et al. 2009). The SLC19A2 gene was first identified in a group of Iranian families affected by a thiamine-responsive megaloblastic anemia syndrome (TRMA), an autosomal recessive disorder (Guerrini et al. 2009).
The SLC19A3 is a second thiamine transporter that has been recently mapped to chromosome 2q37. The murine SLC19A3 gene has also been mapped to chromosome 1 in a seizure susceptibility locus in the DBA/2J mouse strain. In 2005, Zeng and colleagues identified two missense mutations in the SLC19A3 in a group of highly inbred families affected by the biotin-responsive basal ganglia disease (Guerrini et al. 2009). Kono and colleagues (2009) described two cases of familial Wernicke’s encephalopathy caused by two different mutations in the SLC19A3 (K44E and E320Q) responsible for a reduction of intracellular trafficking and thiamine transporter activity. The authors raised the important issue of the responsiveness to thiamine treatment and efficacy of high dose of thiamine for the prophylaxis of the familial Wernicke’s encephalopathy (Kono et al. 2009).
The concept of an abnormal function of one of the thiamine transporters has important implications for the pharmacological treatment of both Wernicke’s encephalopathy and Korsakoff syndrome.
Recently Yamada and colleagues identified a homozygous mutation in exon 3 of SLC19A3 (E320Q) in a Japanese pedigree of four patients who presented with epileptic spasms in early infancy, severe psychomotor retardation and MRI evidence of progressive brain atrophy and bilateral lesions in the thalami and basal ganglia (Yamada et al. 2010). The authors concluded that their cases broadened the phenotypic spectrum of disorders associated with SLC19A3, and highlighted the potential of treatment with thiamine and the importance of assessing therapeutic outcomes.
Our laboratory at University College London (UK) screened the high affinity Thiamine Transporter gene, SLC19A2 on chromosome 1q23.2-q23.3 and the low affinity thiamine transporter gene SLC19A3 on chromosome 2q36.3 in a group of alcoholics affected by WKS and in “super-normal” controls who had been screened not only for a negative history of psychiatric psychopathology but also for a negative family history of psychiatric problems including addictions. We identified several genetic variants in the SLC19A2 (exon 6, 3’ UTR region) and in the SLC19A3 (promoter and intron 1) (Guerrini et al. 2005, 2009). Two of the changes we found in the 3 prime untranslated region of the thiamine transporter gene can potentially affect gene expression. A genetic association study carried out in our laboratory in 109 cases of WKS and 220 normal controls showed evidence of association with four markers in the SLC19A3 gene (p = 0.019-0.039). (Guerrini, unpublished data)
The Wernicke Korsakoff syndrome may be a very complex, multifactorial disorder in the pathogenesis of which the interaction of multiple genes and environment plays an important role. However it is still plausible that “megaphenic” gene effects are responsible for WKS susceptibility (“megaphenic” in the sense of having a large effect on phenotypic variability). The thiamine transport genes are good candidates for having such a role. Further genetic studies are needed to investigate association with candidate genes or linkage with hot spot areas. Furthermore the relationship between the phenotypes highlighted by the studies on the biotin responsive basal ganglia disease and on the familial Wernicke’s encephalopathy and the response to treatment need to be further investigated.
The Dose of Thiamine Required to Treat Wernicke’s Encephalopathy Successfully
The brain weighs approximately 1.5 kg which is only 2 % of the body weight, but receives 15 % of the cardiac output and uses 20 % of the total oxygen consumption of the body. Glucose provides almost all of the fuel in the resting state. About half of the cerebral thiamine (mostly TDP) is involved in pyruvate oxidation and accounts for nearly all of the brain’s oxygen consumption (McIlwain and Bachelard 1971).
The brain requires a continuous supply of thiamine for metabolic and synthetic purposes. At normal plasma concentrations of thiamine, influx into the brain is almost entirely due to a high affinity carrier mediated process. The uptake occurs at 0.3 ug/h/g tissue (Greenwood et al. 1980; Greenwood and Pratt 1982) and is very similar to the rate of thiamine turnover in the brain (0.3-0.5 ug/h/g tissue (Rindi et al. 1980). It is just sufficient to meet the brain’s requirements, making the brain very vulnerable to any fall in circulating levels of thiamine. Passive diffusion can occur but requires high concentrations of thiamine probably not achieved by oral therapy in a malnourished alcoholic patient (Thomson et al. 2002). The requirement for thiamine will be increased if damage has occurred to the apoenzymes and may be a contributory factor to the development of WE and KS (Pratt et al. 1985; Heap et al. 2002). It is also possible that so-called “normal” circulating levels of thiamine may not be adequate if the transport system is genetically abnormal and the enzyme systems within the neurone are damaged. That may explain, at least in part, why alcoholic patients with WE require such large parenteral doses of thiamine.
Current Treatment for Wernicke’s Encephalopathy
De Wardener and Lennox (1947), reporting on Japanese prisoners of war who developed WE, were only able to give an average dose of 2 mg of thiamine by injection (the largest single dose given was 9 mg), together with Japanese tablets of compressed yeast and marmite, because supplies were very limited. Most of their patients survived on these small doses of thiamine and eventually the mental symptoms resolved although some were left with a permanent amnesia for the time during which they were most confused. Of the 31 non-fatal cases, 29 were treated and recovered. Of the 25 receiving intramuscular (IM) thiamine, 20 recovered dramatically within 48 h. Loss of memory for recent events in non-complicated cases typically returned within 2 days to 1 week. During recovery large rice meals would often bring about a temporary relapse of symptoms. The WE in this series was caused by malnutrition alone and no alcohol was consumed. Under these circumstances adequate thiamine (adequate in a therapeutic sense) was available from the small oral and parenteral doses given. This is in contrast to the much higher doses of parenteral thiamine required to treat some patients with WE associated with alcohol misuse (requiring up to 1 g in the first 24 h), presumably where the thiamine transport systems have been damaged.
There are no formal dose-ranging placebo-controlled studies on the use of B-complex vitamins in the treatment of WE. Knowledge of the dose is based on uncontrolled trials and empirical practice. One randomised-controlled trial has assessed the effect of variable thiamine dosing regimes on working memory in alcohol dependent individuals (n = 107; thiamine 5 mg; 20 mg; 50 mg; 100 mg; 200 mg IM daily for 2 days). The 200 mg dose was found to be significantly more effective than the 5 mg dose (Ambrose et al. 2001; Day et al. 2008).
The clinical evaluation of patients at risk of thiamine deficiency is set out in Table 2.
Treatment recommendations for WE have been determined from the following evidence derived from case reports. Cases of WE have been described in patients taking high doses of oral thiamine (Cook et al. 1998). Doses of parenteral thiamine between 100 mg–250 mg do not always prevent death, and between 56 %–84 % of patients with WE are found to develop KS if followed up long-term (Victor et al. 1971; Wood et al. 1986; Thomson et al. 2008). This poor outcome is not necessarily due to irreversible brain damage having been present at the time of presentation. Other studies show that these doses are sub-optimal and may not restore vitamin status, or improve clinical signs or prevent death (Cook et al. 1998). There are case reports of patients requiring up to 1 g of thiamine in the first hours to achieve a clinical response (Cook et al. 1998; Nakada and Knight 1984; Lindberg and Oyler 1990).
The doses of thiamine in Table 3 are recommended by the British National Formulary and the Royal College of Physicians, London (Royal College of Physicians 2001) and have been licensed for use in the UK by the Medicines and Healthcare Products Regulatory Agency (MHRA) since 1994. They are also in accordance with the evidence-based guidelines published by the British Association for Psychopharmacology (Lingford-Hughes et al. 2004), EFNS (Galvin et al. 2010) and NICE guidelines (NICE 2011). Charness (2006) has suggested that these recommendations should be considered for adoption in the United States.
Identification of Patients at Risk of WE
Could careful clinical examination identify alcoholic patients at risk of developing WE? Pitel et al. (2011) used operational criteria developed by Caine et al. (1997) to identify individuals at risk for WE from a sample of 56 abstinent alcoholics. A control group of 38 non alcohol-dependent volunteers also participated. Of the alcoholic group 15 (27 %) met none of the Caine et al. criteria; 32 (57 %) met one criterion and 9 (16 %) met 2 or more criteria. When neuropsychological performance was compared across the three alcoholic sub-groups a graded effect emerged. The alcoholics who met none of the Caine criteria performed as well as the non-alcoholic control group; those meeting one criterion showed mild-to-moderate deficits and those meeting two or more criteria showed the most impairment. Whole blood thiamine diphosphate (TDP) was measured in 28 of the alcoholic group and 22 of the control group. Although there were no significant differences in TDP levels between the alcoholic and control groups, TDP levels were selectively related to memory performance in the alcoholics. This paper has come to some very important conclusions. It has shown that the Caine et al. criteria, originally used to identify patients who were subsequently found to have Wernicke lesions at autopsy, can be applied with advantage to alcoholic groups at risk of developing WE (Pitel et al. 2011).
These finding confirm the importance of maintaining an adequate level of circulating thiamine and that individuals with impaired thiamine transport are at risk of developing memory deficits. This has practical implications for the consideration of thiamine prophylaxis in alcoholics presenting to clinical services, as distinct from specific treatment for Wernicke episodes. Additional clinical indications for thiamine deficiency will be discussed further in the treatment section.
Two recent reviews have emphasized the need to determine the optimum dose of parenteral thiamine for the prophylaxis and treatment of Wernicke’s Encephalopathy (Day et al. 2008; Sechi and Serra 2007). Galvin et al. (2010) compared the clinical features of WE in recent alcoholic and non-alcoholic case series where Caine et al.’s (1997) operational criteria for the identification of WE could be used and MRI studies had been carried out. Dietary deficiency and vomiting were more frequent in non-alcoholics whereas eye and cerebellar signs, the classical triad and altered mental state were more frequently observed in the alcoholics. It is likely that WE secondary to dietary deficiency presents as a more “dramatic acute syndrome” whereas in alcoholics it is a more covert subclinical syndrome which can be missed or mistaken for the effects of alcohol, and thus goes untreated.
It is very difficult to ‘stage’ the extent of damage in the living brain and the degree to which neuropathological impairments correlate with cognitive function. If it can be shown that abnormalities in neural connectivity are associated with deficits in cognitive function then this could afford insights into the progression of WE and potentially act as a marker for recovery. (It is not fully known what neurological processes underlie functional connectivity).
Irreversible brain damage may be present before treatment commences but this cannot be presumed until the optimum dose of thiamine has been determined and given parenterally for an adequate period of time with long-term follow-up of the patient. At the present time it is not known what that dose is for every patient. In the UK parenteral thiamine is given as Pabrinex, a parenteral formulation containing 500 mg ascorbic acid, 160 mg nicotinamide, 50 mg pyridoxine hydrochloride, 4 mg riboflavin and 250 mg of thiamine hydrochloride. This treatment has the advantage of replacing a number of nutrients which may be deficient, especially nicotinic acid.
Recent studies carried out in the UK have shown that many hospitals do not have guidelines for treating WE, and where they exist, they are not always followed by medical staff (Littlewood et al. 2008).
In reviewing treatment studies it should be recognised that while “alcoholics” are used as subjects, consideration of the following is often not taken into account:
-
Duration and severity of alcohol dependence and the duration of periods of abstinence;
-
Pattern of drinking e.g. binge drinking;
-
Gender (most alcohol studies involve men); young women with concurrent binge eating and drinking disorders may be particularly at risk;
-
Age of onset of drinking and dependent drinking (individuals with a genetic predisposition are usually alcohol dependent by the age of 25 years);
-
Onset and severity of complications such as alcohol withdrawal seizures and DTs;
-
Onset and severity of physical complications such as alcohol-related liver disease;
-
Body Mass Index and other measures of nutrition/malnutrition (especially in young women);
-
History of episodes of alcohol detoxification (number, quality, whether parenteral thiamine previously given).
It is possible that the effect of alcohol on the brain (alcohol-induced neurotoxicity) modulates the response to treatment with thiamine. Younger people may present in extreme circumstances but are likely to respond more quickly to thiamine replacement whereas older people with a more chronic alcohol use disorder and multiple subclinical WE episodes are at risk of poorer outcomes.
Clinical Aspects of Korsakoff’s Syndrome
KS is characterised by a profound anterograde and retrograde amnesia, disorientation in place and time and lack of insight. Peripheral neuropathy, nystagmus and ataxia may also be present, the latter two signs being related to earlier Wernicke episodes. Other common signs and symptoms include confabulation, anxiety and apathy. Anterograde amnesia is an impaired ability to acquire new episodic memories, for instance when or where an event took place, which helps to reconstruct the source of the event.
KS patients perform poorly in tests of frontal lobe function, particularly in tests of executive functioning (Kopelman 1989) and impairments have also been shown in spatiotemporal processing (Kopelman 1989). IQ is often intact as is working memory (Jacobson and Lishman 1987) and procedural and semantic memory may also be preserved. Recent work supports the theory that ARBD deficits exist along a continuum (Pitel et al. 2008).
The clinical assessment of individuals with KS requires attention to history taking and physical examination (Kopelman et al. 2009). A collateral history from family or friends will help determine the premorbid functioning of the patient before admission to hospital, their alcohol use history and complications, and the observed decline in functioning. Physical examination may uncover peripheral neuropathy and cerebellar ataxia. Assessment of cognitive function should not be carried out until at least 4 weeks post alcohol withdrawal.
Services
Patients with KS and ARBD require well-integrated support in a managed care network. In the UK there are examples of best practice in Scotland and North West England (Thomson et al. 2012), but such multi-disciplinary services are the exception rather that the rule. Provision could take the form of a brain injury rehabilitation service, or, as in Glasgow, consist of a designated community team with a residential component.
As well as guidance on treatment with thiamine, consideration needs to be given to formal treatment packages which should aim to maintain abstinence in a supported housing/rehabilitation environment, as well as ongoing follow-up, to monitor any improvements in cognitive status, and psychiatric and functional well-being (Leenane 1986). The needs of these individuals are likely to be wide-ranging and variable. It may be possible to support some in their own homes, at least in the initial stages. Others will require life-long residential support from the moment they receive the diagnosis. In this situation it should be ensured that patients have their own rooms which they can personalise.
Abstinence can lead to and facilitate improvement. Families and care-givers will need to be educated and supported. These patients are younger than the typical dementia population so are physically more active and can become very frustrated, suspicious, angry and aggressive in residential care settings. They often query why they are living in such a setting and repeatedly request to return home. The challenge is to allow them some “independence” in the “cared-for” setting, while at the same time meeting their needs and keeping them safe. An individually-tailored cognitive behavioural approach will allow the development of coping strategies for dealing with day-to-day routines. The writing down of information and breaking down of activities into component steps (distributive practice) and the use of diaries, lists or prompts can all help. Errorless learning has a major role to play in the management of this patient group.
Conclusions
KS occurs because WE is misdiagnosed or its treatment with thiamine is inadequate. Sometimes patients have already sustained permanent or semi-permanent brain “injury” by the time they present, and this can be a limiting factor in the response to treatment. It therefore follows that patients at risk must be identified and treated with an adequate amount of parenteral thiamine. Timely intervention is critical, before the changes occurring in the brain increase the requirement for thiamine beyond what the patient is capable of supplying.
It has been suggested that there is a difference between WKS due to dietary deficiency alone and WKS related to thiamine deficiency and alcohol misuse. The evidence rests on the fact that KS rarely supervenes in the former situation but frequently occurs in alcoholic patients. That the underlying mechanism is different is also indicated by the higher doses of thiamine required to treat WE in the alcohol group.
These two causes of WE are likely to be very different. In the case of WE due to an inadequate intake of dietary thiamine, the biochemical consequences which follow are relatively simple. This is in contrast to the extensive changes within the body and brain induced by a combination of chronic alcohol misuse (dependence) and thiamine deficiency. In the latter situation larger doses of parenteral thiamine are required. However replacement of thiamine alone will not automatically and immediately repair the extensive damage which has been sustained and such changes are likely to persist well beyond the initial replacement. It is therefore important to understand the biochemical changes induced and the interference with the absorption of vitamins so that they can be reversed before WE occurs and to investigate whether they may contribute to the brain damage associated with KS.
It is important to take into account the fact that normal levels of circulating thiamine diphosphate or transketolase activity in the red blood cells do not guarantee normal thiamine diphosphate or thiamine triphosphate levels in the brain cells because of possible transport problems into the neurone. Furthermore, measuring one enzyme activity does not mean that all of the enzymes are working normally. The requirement for thiamine is determined to some extent by damage to the apoenzyme caused by alcohol metabolism and the effect on the thiamine transport system.
We are faced with the reality that many alcohol dependent patients with WE develop KS even after treatment with thiamine. This is sometimes due to the fact that the thiamine has been given orally and it is impossible for the patient to absorb adequate amounts of thiamine to develop a blood concentration sufficient to pass the blood brain barrier and supply the brain cells with adequate amounts of thiamine. Some individuals are at increased risk because of a genetic predisposition, which means that their thiamine requirements are increased, and higher blood concentrations are required for thiamine to enter the brain cells. Sometimes patients have already sustained irreversible brain damage at the time of presentation.
The response to parenteral thiamine is variable in different patients, with some responding to relatively low doses of thiamine but others requiring more than 1 g of thiamine parenterally in the first 24 h. Unfortunately there are no formal dose-ranging placebo-controlled studies to guide treatment, either in relation to the required dose in any specific patient, nor in relation to how long and at what dose the treatment should be continued. Although replacement of thiamine will allow the brain cells to respond, to some extent this response will be limited by other nutrient deficiencies, e.g., folate, magnesium and it will take time to repair the damage done by neurotoxicity due to the combined effects of alcohol misuse and thiamine deficiency. The logical response to this dilemma is to give a continuing dose of high level parenteral thiamine while supplying all of the other nutrients required for brain function and to monitor the patient’s progress, if necessary, for a number of months. Ongoing abstinence and a corrective diet are probably required to optimise thiamine replacement.
Little research has been carried out into the causes of alcohol-related brain damage other than thiamine. As well as thiamine deficiency, future research should explore the synergistic effects of general nutritional status, and of repeated episodes of alcohol intoxication and withdrawal on the alcoholic brain.
Identification, early intervention and appropriate treatment with thiamine, abstinence from alcohol and integrated and flexible services are all needed to meet the needs of these stigmatised and marginalised patients.
References
Acker, W., Aps, E. J., Majumdar, S. K., Shaw, G. K., & Thomson, A. D. (1982). The relationship between brain and liver damage in chronic alcoholic patients. Journal of Neurology, Neurosurgery and Psychiatry, 45, 984–987.
Ambrose, M. L., Bowden, S. C., & Whelan, G. (2001). Thiamin treatment and working memory function of alcohol-dependent people: preliminary findings. Alcohol: Clinical and Experimental Research, 25, 112–116.
Baker, H., Thomson, A. D., & Frank, O. (1974). Absorption and passage of fat and water soluble thiamine derivatives into erythrocytes and cerebro-spinal fluid in man. American Journal of Clinical Nutrition, 27, 676–680.
Brust, J. C. M. (2010). Ethanol and cognition: indirect effects, neurotoxicity and neuroprotection: a review. International Journal of Environmental Research Public Health, 7, 1540–1557.
Butterworth, R. F., Kril, J. J., & Harper, C. (1993). Thiamine-dependent enzyme changes in brain of alcoholics: relationship to Wernicke–Korsakoff syndrome. Alcoholism, Clinical and Experimental Research, 17, 1084–8.
Caine, D., Halliday, G. M., Krill, J. J., & Harper, C. G. (1997). Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s Encephalopathy. Journal of Neurology, Neurosurgery & Psychiatry, 62, 51–60.
Charness, M. E. (2006). Alcohol. In J. Noseworthy (Ed.), Neurological therapeutics (pp. 1677–1685). London: Martin Dunitz.
Ciccia, R. M., & Langlais, P. J. (2000). An examination of the synergistic interaction of ethanol and thiamine deficiency in the development of neurological signs and long term cognitive and memory impairments. Alcoholism, Clinical and Experimental Research, 24, 622–634.
Cook, C. C., Hallwood, P. M., & Thomson, A. D. (1998). B-vitamin deficiency and neuro-psychiatric syndromes in alcohol misuse. Alcohol and Alcoholism, 33, 317–316.
Crowe, S. F., & Kempton, S. (1997). Both ethanol toxicity and thiamine deficiency are necessary to produce long-term memory deficits in the young chick. Pharmacology, Biochemistry and Behaviour, 58, 461–70.
Crowe, S. F., & El-Hadj, D. (2002). Phenytoin ameliorates the memory defect induced in the young chick by ethanol toxicity in association with thiamine deficiency. Pharmacology, Biochemistry and Behaviour, 71, 215–221.
Day, E., Bentham, P., Callaghan, R., et al. (2008). Thiamine for Wernicke–Korsakoff Syndrome in people at risk from alcohol abuse (review). The Cochrane Collaboration. Chichester: Wiley.
De Wardener, H. E., & Lennox, B. (1947). Cerebral beriberi (Wernicke's Encephalopathy): review of 52 cases in a Singapore prisoner-of-war hospital. Lancet, 1, 11–17.
Diaz, G. A., Banikazemi, M., Oishi, K., Desnick, R. J., & Gelb, B. D. (1999). Mutations in a new gene encoding a thiamine transporter cause thiamine- responsive megaloblastic anaemia syndrome. Nature Genet, 22, 309–12.
Flink, E. B. (1969). Therapy of magnesium deficiency. Ann NY Academy of Science, 162, 901–905.
Galvin, R., Bråthen, G., Ivashynka, A., Hillbom, M., Tanasescu, R., & Leone, M. A. (2010). EFNS guidelines for diagnosis, therapy and prevention of Wernicke Encephalopathy. European Journal of Neurology, 17, 1408–1418.
Gilchrist, G., & Morrison, D. (2005). Prevalence of alcohol-related brain damage among homeless hostel dwellers in Glasgow. European Journal of Public Health, 15, 587–588.
Greenwood, J., & Pratt, O. E. (1982). Inhibition of thiamine transport across the blood–brain barrier in the rat by chemical analogue of the vitamin. The Journal of Physiology, 336, 479–486.
Greenwood, J., Love, E. R., & Pratt, O. E. (1980). Carrier mediated transport of thiamine across the blood–brain barrier. Proceedings of the Physiological Society, Journal of Physiology, 310, 23.
Guerrini, I., Thomson, A. D., Cook, C. C. H., et al. (2004). Direct genomic sequencing of the high affinity thiamine transporter gene and the OGDH gene in alcoholics with Wernicke-Korsakoff Syndrome. Am J Med Genetics (Neuropsychiatric Genetics), 130B, 17–18.
Guerrini, I., Thomson, A. D., Cook, C. C., McQuillin, A., Sharma, V., Kopelman, M., Reynolds, G., Jauhar, P., Harper, C., & Gurling, H. M. (2005). Direct genomic PCR sequencing of the high affinity thiamine transporter (SLC19A2) gene identifies three genetic variants in Wernicke Korsakoff syndrome (WKS). AmJ Med Genet B Neuropsychiatr Genet, 137, 17–19.
Guerrini, I., Thomson, A. D., & Gurling, H. M. (2009). Molecular genetics of alcohol related brain damage. Alcohol and Alcoholism, 44, 166–170.
Harper, C. (2006). Thiamine (vitamin B1) deficiency and associated brain damage is still common throughout the world and prevention is simple and safe. European Journal of Neurology, 13, 1–6.
Harper, C. G., Sheedy, D. L., Lara, A. I., et al. (1988). Prevalence of Wernicke–Korsakoff syndrome in Australia: Has thiamine fortification made a difference? The Medical Journal of Australia, 168, 542–5.
Harper, C., Dixon, G., Sheedy, D., et al. (2003). Neuropathological alterations in alcoholic brains. Studies arising from the New South Wales Tissue Resource Centre. Prog Neuropsychopharmacol Biol Psychiatry, 27, 951–61.
Heap, L. C., Pratt, O. E., Ward, R. J., et al. (2002). Individual susceptibility to Wernicke–Korsakoff syndrome and alcoholism induced cognitive deficit: impaired thiamine utilization found in alcoholics and alcohol abusers. Psychiatric Genetics, 12, 217–224.
Hoyumpa, A. M., Nichols, S., Schenker, S., & Wilson, F. N. (1976). Thiamine transport in thiamine-deficient rats. Biochem Biophys Acta, 436, 438.
Howard, L. C., Wagner, C., & Schenker, S. (1974). Malabsorption of thiamine in folate deficient rats. Journal of Nutrition, 104, 1024.
Jacobson, R. R., & Lishman, W. A. (1987). Selective memory loss and global intellectual deficits in alcoholic Korsakoff’s syndrome. Psychological Medicine, 17, 649–655.
Keedwell, P. A., Poon, L., Papadopoulos, A. S., Marshall, E. J., & Checkley, S. (2001). Salivary cortisol during a medically assisted alcohol withdrawal. Addiction Biology, 6, 247–257.
Kono, S., Miyajima, H., Yoshida, K., Togawa, A., Shirakawa, K., & Suzuki, H. (2009). Mutations in a thiamine-transporter gene and Wernicke's-like encephalopathy. The New England Journal of Medicine, 360, 1792–4.
Kopelman, M. (1989). Remote and autobiographical memory, temporal context memory and frontal atrophy in Korsakoff and Alzheimer patienta. Neuropsychologica, 27, 437–460.
Kopelman, M. D., Thomson, A. D., Guerrini, I., & Marshall, E. J. (2009). The Korsakoff syndrome: clinical aspects, psychology and treatment. Alcohol and Alcoholism, 44, 148–154.
Leenane, K. J. (1986). Management of moderate to severe alcohol-related brain damage (Korsakoff’s Syndrome). Medical Journal of Australia, 145, 136–143.
Leevy, C. M., Cardi, L., Frank, O., Gellene, R., & Baker, H. (1965). Incidence and significance of hypovitaminemia in a randomly selected municipal hospital population. American Journal of Clinical Nutrition, 17, 259–271.
Lieber, C. S., & De Carli, L. M. (1989). Liquid diet technique of ethanol administration: 1989 update. Alcohol and Alcoholism, 24, 197–211.
Lindberg, M. C., & Oyler, R. A. (1990). Wernicke’s Encephalopathy. American Family Physician, 41, 1205–1209.
Lingford-Hughes, A. R., Welch, S., & Nutt, D. J. (2004). Evidence based guidelines for the pharmacological management of substance misuse, addiction and comorbidity: recommendations from the British Association for Psychopharmacology. J Psychopharmacology, 18, 293–335.
Lishman, W. A. (1990). Alcohol and the brain. The British Journal of Psychiatry, 156, 635–644.
Littlewood NK, McWhirter K, Mcnaughton G (2008) Pabrinex® prescribing in Scottish emergency departments. Poster presented at the Inaugural Scientific Conference of The College of Emergency Medicine, London, UK; 14–16 May 2008
MacRae, S., & Cox, S. (2003). Meeting the needs of people with alcohol related brain damage: a literature review on the existing and recommended service provision and models of care. Sterling: University of Sterling.
McIlwain, H., & Bachelard, H. (1971). Biochemistry and the nervous system, 4th edn (pp 226–307). Edinburgh: Churchill Livingstone.
Mancinelli, R., Ceccani, M., Guiducci, M. S., et al. (2003). Simultaneous liquid chromatography assessment of thiamine, thiamine monophosphate and thiamine diphosphate in human erythrocytes: a study on alcoholics. Journal of Chromatography, 789, 355–363.
Marshall, E. J., Humphreys, K., & Ball, D. M. (2010). The treatment of drinking problems (5th ed.). Cambridge: Cambridge University Press.
Matsumoto, I. (2009). Proteomics approach in the study of the pathophysiology of alcohol-related brain damage. Alcohol and Alcoholism, 44, 171–176.
Mendelson, J. H., & Stein, S. (1966). Serum cortisol levels in alcoholic and non-alcoholic subjects during experimentally induced ethanol intoxication. Psychosomatic Medicine, 28, 212–226.
Nakada, T., & Knight, R. T. (1984). Alcohol and the central nervous system. The Medical Clinics of North America, 68, 121–131.
NICE (2011). Alcohol-use disorders: diagnosis, assessment and management of harmful drinking and alcohol dependence. Clinical Guideline 115. London: NICE.
Nishino, K., & Itokawa, Y. (1977). Thiamine metabolism in vitamin B6 and B12 deficient rats. Nutrition, 107, 775.
Pitel, A.-L., Beaunieux, H., Witkowski, T., Vabret, F., de la Sayette, V., Viader, F., Desgranges, B., & Eustache, F. (2008). Episodic and working memory deficits in alcoholic Korsakoff patients: the continuity theory revisited. Alcohol: Clinical and Experimental Research, 32, 1229–1241.
Pitel, A.-L., Zahr, N. M., Jackson, K., Sassoon, S. A., Rosenbloom, M. J., Pfefferbaum, A., & Sullivan, E. V. (2011). Signs of preclinical Wernicke’s encephalopathy and thiamine levels as predictors of neuropsychological deficits in alcoholism without Korsakoff’s Syndrome. Neuropsychopharmacology, 36, 580–588.
Pratt, O. E., Jeyasingham, M., Shaw, G. K., et al. (1985). Transketolase variant enzymes and brain damage. Alcohol and Alcoholism, 20, 223–232.
Price, J., Mitchell, S., Wiltshire, B., Graham, J., & Williams, G. (1988). A follow-up study of patients with alcohol-related brain damage in the community. Australian Drug and Alcohol Review, 7, 83–87.
Price, J., Hicks, M., & Williams, G. (1993). A feature of alcoholic Wernicke’s encephalopathy favourable to the maintenance of memory function: vomiting. Alcohol and Alcoholism, 28, 339–345.
Ramayya, A., & Jauhar, P. (1997). Increasing incidence of Korsakoff’s psychosis in the East End of Glasgow. Alcohol and Alcoholism, 32, 281–285.
Rindi, G., Patrini, C., Comincioli, V., & Reggiani, C. (1980). Thiamine content and turnover rates of some rat nervous regions, using labeled thiamine as a tracer. Brain Research, 81, 369–80.
Royal College of Physicians. (2001). Alcohol-can the NHS afford it? Recommendations for a coherent alcohol strategy for hospitals. London: Royal College of Physicians.
Russell, C., & Elia, M. (2009). Nutrition screening survey in the UK in 2008: hospitals, care homes and mental health units. Redditch: British Association for Parenteral and Enteral Nutrition.
Sechi, G., & Serra, A. (2007). Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurology, 6, 442–455.
Subramanian, V. S., Subramanya, S. B., Tsukamoto, H., & Said, H. M. (2010). Effect of chronic alcohol feeding of physiological and molecular parameters of renal thiamine transport. American Journal of Physiology – Renal Physiology, 299(1), F28–F34.
Subramanya, S. B., Subramanian, V. S., & Said, H. M. (2010). Chronic alcohol consumption and intestinal thiamin absorption: effects on physiological and molecular parameters of the uptake process. American Journal of Physiology – Gastrointestinal and Liver Physiology, 299(1), G23–G31.
Thomson, A. D. (1966). The absorption of sulphur-labelled thiamine hydrochloride in control subjects and in patients with intestinal malabsorption. Clinical Science, 31(2), 167–179.
Thomson, A. D. (2000). Mechanisms of vitamin deficiency in chronic alcohol misusers and the development of the Wernicke-Korsakoff Syndrome. Alcohol and Alcoholism, 35(Suppl. 1), 2–7.
Thomson, A. D., & Leevy, C. M. (1972). Observations on the mechanism of thiamine hydrochloride absorption in man. Clinical Science, 43, 153.
Thomson, A. D., & Marshall, E. J. (2006). The treatment of patients at risk of developing Wernicke's encephalopathy in the community. Alcohol and Alcoholism, 41, 159–167.
Thomson, A. D., Baker, H., & Leevy, C. M. (1968). Thiamine absorption in alcoholism. American Journal of Clinical Nutrition, 21, 537.
Thomson, A. D., Baker, H., & Leevy, C. M. (1970). Patterns of 35S-thiamine hydrochloride absorption in the malnourished alcoholic. Journal of Laboratory and Clinical Medicine, 76(1), 34–45.
Thomson, A. D., Frank, O., Baker, H., et al. (1971). Thiamine propyl disulfide: absorption and utilization. Annals of Internal Medicine, 74, 529–534.
Thomson, A. D., Frank, O., De Angelis, B., & Baker, H. (1972). Thiamine depletion induced by dietary folate deficiency in rats. Nutrition Reports International, 6, 107–110.
Thomson, A. D., Ryle, P. R., & Shaw, G. K. (1983). Ethanol, thiamine and brain damage. Alcohol and Alcoholism, 18, 27–43.
Thomson, A. D., Jeyasingham, M., Pratt, O. E., et al. (1987). Nutrition and alcoholic encephalopathies. Acta Medica Scandinavica. Supplementum, 717, 55–65.
Thomson, A. D., Cook, C. C. H., Touquet, R., et al. (2002). The Royal College of Physicians report on alcohol: guidelines for managing Wernicke’s encephalopathy in the Accident and Emergency Department. Alcohol and Alcoholism, 37, 513–521.
Thomson, A. D., Cook, C. C. H., Guerrini, I., et al. (2008). Wernicke’s Encephalopathy: plus ca change, plus c’est la meme chose. Alcohol and Alcoholism, 43, 180–186.
Thomson, A. D., Guerrini, I., & Marshall, J. E. (2009). Wernicke’s encephalopathy: role of thiamine. Practical Gastroenterol, XXXIII(6), 21–30.
Thomson, A. D., Guerrini, I., Bell, D., Drummond, C., Duka, T., Field, M., Kopelman, M., Lingford-Hughes, A., Smith, I., Wilson, K., & Marshall, E. J. (2012). Alcohol related brain damage: report from a medical council on alcohol symposium, June 2010. Alcohol Alcoholism, 47(2), 84–91.
Torvik, A., Lindboe, C. F., & Rogde, S. (1982). Brain lesions in alcoholics. A neuopathological study with clinical correlations. Journal of Neurological Sciences, 56, 233–248.
Victor, M., & Adams, R. D. (1985). The alcoholic dementias. In P. J. Vinken, G. W. Bruyn, & H. L. Klawans (Eds.), Handbook of clinical neurology (Vol 46, pp. 335–353). Amsterdam: Elsevier.
Victor, M., Adams, R. D., & Collins, G. H. (1971). The Wernicke–Korsakoff Syndrome. Philadelphia, PA: F.A. Davis.
Victor, M., Adams, R. D., & Collins, G. H. (1989). The Wernicke–Korsakoff Syndrome and related neurological disorders due to alcoholism and malnutrition. Philadelphia, PA: F.A. Davis.
Witt, E. D., & Goldman-Rakic, P. S. (1983a). Intermittent thiamine deficiency in the rhesus monkey. 1. Progression of neurological signs and neuroanatomical lesions. Annals of Neurology, 13, 376–395.
Witt, E. D., & Goldman-Rakic, P. S. (1983b). Intermittent thiamine deficiency in the rhesus monkey. II Evidence for memory loss. Ann Neurol, 13, 396–401.
Wood, B. (1985). Thiamin status in Australia. World Review of Nutrition and Dietetics, 46, 148–218.
Wood, B., Currie, J., & Breen, K. (1986). Wernicke’s encephalopathy in a metropolitan hospital: a prospective study of the incidence, characteristics and outcome. Medical Journal of Australia, 144, 12–16.
Yamada, K., Miura, K., Hara, K., Suzuki, M., Nakanishi, K., Kumagai, T., Ishihara, N., Yamada, Y., Kuwano, R., Tsuji, S., & Wakamatsu, N. (2010). A wide spectrum of clinical and brain MRI findings in patients with SLC19A3 mutations. BMC Medical Genetics, 11, 171.
Zahr, N., Kaufman, K. L., & Harper, C. G. (2011). Clinical and pathological features of alcohol-related brain damage. Nature Reviews Neurology, 7, 284–294.
Zeng, W. Q., Al-Yamani, E., Acierno, J. S., Jr., Slaugenhaupt, S., Gillis, T., MacDonald, M. E., Ozand, P. T., & Gusella, J. F. (2005). Biotin-responsive basal ganglia disease maps to 2q36.3 and is due to mutations in SLC19A3. American Journal of Human Genetics, 77, 16–26.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Thomson, A.D., Guerrini, I. & Marshall, E.J. The Evolution and Treatment of Korsakoff's Syndrome. Neuropsychol Rev 22, 81–92 (2012). https://doi.org/10.1007/s11065-012-9196-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11065-012-9196-z