
Abstract
Toll-like receptors (TLRs), well-characterized pattern-recognizing receptors of the innate arm of the immune system, are vital in detecting pathogen-associated molecular patterns (PAMPs). The TLR-PAMP interaction initiates an intracellular signaling cascade, predominantly culminating in upregulation of antiviral components, including inducible nitric oxide synthase (iNOS). After activation, various TLR pathways can promote iNOS production via the myeloid differentiation primary response-88 (MyD-88) adapter protein. Subsequently, iNOS facilitates production of nitric oxide (NO), a highly reactive and potent antiviral molecule that can inhibit replication of RNA and DNA viruses. Furthermore, NO can diffuse freely across cell membranes and elicit antiviral mechanisms in various ways, including direct and indirect damage to viral genomes. This review emphasizes current knowledge of NO-mediated antiviral responses elicited after activation of TLR signaling pathways.
Similar content being viewed by others

Introduction
The innate immune system, which mounts host responses against invading pathogens, is equipped with a broad range of germ-line-encoded host receptors referred to as pattern-recognition receptors (PRRs), including NOD-like receptors (NLRs), toll-like receptors (TLRs), RIG-like receptors (RLRs), and C-lectin-type receptors (CLRs) [94]. These innate receptors are capable of recognizing microbial pathogens (e.g., viruses, bacteria and fungi) due to the presence of pathogen-associated molecular patterns (PAMPs), molecules that are highly conserved among microbes. Of the aforementioned PRRs, TLRs are well characterized and indispensable in detecting PAMPs of viruses and other pathogens [81]. When a TLR recognizes a PAMP, that activates an intracellular signaling cascade [117], culminating in upregulation of gene transcription for production of innate antiviral components, including type-1 interferons (IFNs) and pro-inflammatory mediators, including inducible nitric oxide synthase (iNOS) [137]. The latter enzyme promotes production of nitric oxide (NO), which can inhibit viral replication, both directly and indirectly [5, 120].
In the last two decades, our understanding of TLR biology has progressed substantially and been the subject of many reviews [1, 64, 117, 135, 137, 140]. However, none of these reviews has focused on TLR-signaling-mediated production of NO leading to antiviral responses. Therefore, the primary purpose of this review is to discuss current knowledge of NO-mediated antiviral responses elicited following activation of TLR signaling pathways.
Toll-like receptors
The first TLR, identified in an insect (Drosophila), was a molecule with a critical role in antifungal responses. Subsequently, 13 types of TLRs (TLR1, TLR2, TLR3, TLR4, TLR5, TLR6, TLR7, TLR8, TLR9, TLR10, TLR11, TLR12 and TLR13) have been identified in mammals. It is noteworthy that TLR1-9 are present in both humans and mice, whereas TLR11, TLR12 and TLR13 are present only in mice (Fig. 1) [117]. Finally, TLR10 was identified in humans; mice have a TLR10 gene, but it is interrupted and nonfunctional [42]. In birds, TLR2a, TLR2b, TLR4, TLR5 and TLR7 are comparable to their counterparts in humans and mice, whereas TLR1La, TLR1Lb and TLR15 are exclusive to birds [17]. Furthermore, in birds, the TLR8 gene is apparently nonfunctional [106] and the TLR9 gene is missing [136, 148]. That notwithstanding, TLR21, which is unique in birds and fish, has functions similar to those of TLR9 in mammals [16].

Common and specific TLRs in humans, mice and birds. In mammals, TLR2 can dimerize with TLR1 or TLR6, whereas in birds, TLR2 is divided into TLR2a and TLR2b and TLR1 is divided into TLR1La and TLR1Lb (due to gene duplication). TLR2s can form a heterodimer complex with TLR1Ls
Expression of TLRs
TLRs are expressed on various immune and non-immune cells, including macrophages, T and B lymphocytes, and epithelial cells, [9, 53]. In addition, cells in muscle, heart, brain and reproductive organs (testis, ovary, uterus and placenta) also express TLRs [38, 91]. That preferential expression of TLR types varies among cell types suggests activation of specific TLR signaling pathways depending on the type of cells involved. For example, most human peripheral blood mononuclear cells (PBMCs) express TLR1 and TLR6 [53], monocytes and B cells preferentially express TLR2 and TLR10, respectively, and B cells and subsets of dendritic cells highly express TLR7 and TLR9 [53]. Furthermore, TLR7 and TLR9 are highly expressed in dendritic precursor cells following stimulation, and precursor cells of monocytes can be stimulated to upregulate TLR2 and TLR4 [58].
Expression of TLRs varies among host cells; they are expressed on the cell membrane, endosomal membrane, or both, depending on the type of TLRs (Fig. 2). For example, TLR1-2, 5-6 and 10-11 are expressed on the cell surface and distinguish PAMPs on the surface of microbes [132], whereas, TLR3, 7, 8, 9 and 21 are expressed intracellularly on the membrane of the endosomal compartment, and they strategically recognize microbial nucleic acid components during replication [7, 47, 48]. Additionally, TLR4 can be expressed on both cellular and endosomal membranes [133] and is capable of interacting with PAMPs that are on the surface of the microbes or are exposed during replication within the host cell.

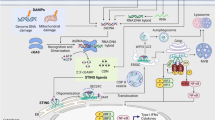
Illustration of potential synthesis of NO via TLR signaling leading to NO-mediated antiviral activity. The TLRs are expressed on the cell surface or inside cells. Among those expressed on the surface, TLR2, 4, 5 and 11 are well studied with respect to iNOS expression. TLR3, 7, 8 and 9/21 are expressed on the membrane of the endosomal compartment and recognize nucleic-acid-based PAMPs, leading to expression of iNOS (among many other mediators). Most of these TLRs use MyD-88 for downstream signaling, but TLR3 uses TRIF protein as an adaptor molecule. In downstream signaling, activated NF-κB or AP-1 enters the nucleus and upregulates gene transcription for iNOS, which facilitates conversion of L-arginine to L-citrulline (using NADPH as an electron donor) to generate highly reactive NO, which has various antiviral effects. TLR, toll-like receptor; LTA, lipoteichoic acid; LPS, lipopolysaccharide; CpG, CpG motif of unmethylated DNA; Poly I:C, polyinosine-polycytidylic acid; MyD-88, myeloid differentiation primary response 88; TRIF, TIR-domain-containing adaptor inducing IFN; IRAKs, IL-1 receptor-associated kinases; TRAF, TNF-receptor-associated factor; TAK1, transforming growth factor beta-activated kinase-1; IKKε, IkappaB kinase-epsilon; RIP1, receptor-interacting protein kinase 1; NF-κB, nuclear factor kappa B; AP-1, activator protein-1; iNOS, inducible nitric oxide synthase; NADPH, nicotinamide adenine dinucleotide phosphate H; NO, nitric oxide
TLR ligands
Each TLR binds to a unique set of ligands (PAMPs of microbes or synthetic compounds) in order to activate signaling pathways. Of the surface-expressing TLRs, TLR2 mainly recognizes the peptidoglycan and lipoteichoic acid (LTA) present in Gram-positive bacteria [66, 138]. Additionally, TLR2 can recognize zymosan, a cell wall component of yeast [119], hemagglutinin protein of measles virus [14], core protein and nonstructural-3 protein of hepatitis C virus [29], and surface glycoproteins (gH, gL and gB) of herpes simplex virus (HSV) [79]. It appears that TLR2 is mainly capable of recognizing lipoproteins or lipopeptides; perhaps at least some of the other putative TLR2 ligands were misclassified due to contamination with highly active natural lipoproteins or lipopeptides [149]. Furthermore, TLR2 can form a heterodimer complex with TLR1 or TLR6 and recognize lipopeptides present in various bacteria [121]. Similarly, TLR1 associated with TLR2 recognizes triacyl lipopeptides, whereas a TLR6-TLR2 complex recognizes diacyl lipopeptides [55]. In addition, TLR4 binds to bacterial endotoxin and lipopolysaccharide (LPS), a component of the cell wall of Gram-negative bacteria [11]. However, for initiation of LPS signaling, TLR4 requires association with another surface molecule, myeloid differentiation factor 2 (MD-2) [124], and CD14 [109]. Moreover, TLR4 can recognize cell wall components of viruses, fungi and helminths, including the envelope protein of murine retroviruses, HIV-1 and human endogenous retrovirus [96, 111, 114], the fusion protein of respiratory syncytial virus [43], α-glucan and mannan of fungus [15, 127], and lacto-N-fucopentaose III of helminths [139]. Expression of TLR5 occurs in intestinal epithelial cells, mainly at the basolateral surface; it recognizes invading flagellated pathogenic bacteria by identifying a protein called flagellin [33]. The amino acid sequence of TLR5 appears to be similar to those of TLR11 and TLR12 [115]; the latter two TLRs form a complex and recognize profilin-like proteins in Toxoplasma gondii (protozoan parasite) [108]. Finally, TLR10 may sense ligands derived from within the host rather than from microbes [102].
Considering endosomal TLRs, TLR3 binds to viral double-stranded RNA (dsRNA) and polyinosinic polycytidylic acid (polyI:C; a synthetic compound, structurally similar to viral dsRNA) [86], whereas TLR7 and 8 bind to viral single-stranded RNA (ssRNA) [47]. Two receptors, TLR9 in mammals and TLR21 in birds, are the only ones known to detect both bacterial and viral DNA containing unmethylated cytosine-guanosine deoxynucleotides (CpG) motifs, which are generally methylated in vertebrate genomes [48]. Consequently, the frequency of CpG motifs is negligible in vertebrate DNA, although it occurs with high frequency in microbial genomes [71]. Differences in methylation and the prevalence of unmethylated CpG motifs in DNA of microbes (bacteria, fungus and viruses) allow selective host responses against DNA of microbial origin. CpG DNA has three major classes (A, B and C) based on structural variations and effects on PBMCs [25]. Class A CpG DNA (also known as ODN 2216) predominantly activates dendritic cells and natural killer (NK) cells, with effects mediated via interferon regulating factor (IRF) 7 signaling pathways from early endosomes that promote production of type 1 IFN. Class B (ODN 2007) is a strong activator of B cells and monocytes and operates via nuclear factor kappa (NF-κ) B signaling pathway from late endosomes, leading to production of pro-inflammatory mediators. Finally, class C CpG DNA has characteristics of both class A and B [72] in terms of both structure and function.
Detailed understanding of TLR structure and signaling mechanisms have enabled development of specific synthetic ligands with therapeutic potential [144]. In that regard, these ligands can be used to manipulate the host immune system [51, 144].
Structure and signaling mechanism of TLRs
Structure of TLRs
The TLRs are in the type 1 transmembrane protein family. Structurally, each TLR expressed on a cell membrane consists of a cytoplasmic C-terminal domain, a transmembrane component, and an N-terminal domain that is exposed to the outside of the cell [37]. In direct contrast, in endosomal TLRs, the N-terminal region is exposed internally and the C-terminal region is exposed externally [55]. The C-terminal domain is conserved and is homologous to the internal domain of an interleukin 1 receptor (IL-1R) named Toll/IL-1R (TIR); therefore, TLRs are classified as a subfamily of the interleukin-1 receptor/toll-like receptor superfamily [100]. Although the N-terminal domain of most TLRs has a horseshoe shape, extracellular regions of TLR8 and TLR9 are ring-shaped [101, 134]. The extracellular component of TLR2 can form a heterodimer with the extracellular domain of TLR1 or TLR6, although other TLRs can form only homodimers [28]. Furthermore, before and after ligand binding, TLR9 is a monomer and a dimer, respectively [101]. Cluster of differentiation-14 (CD-14) molecules can be associated with TLR4, TLR1/TLR2 or TLR2/TLR6 [12], and a concerted effort is required for recognition of certain ligands. Conversely, the curved N-terminal domain of TLRs interacts with ligands, and the ligand binding sites consist of many leucine-rich repeats (LRR). The length and the sequence of LRRs vary among TLRs [20]. In contrast to other TLRs, TLR7,8 and 9 contain a long insertion loop region (Z-loop) of ~40 amino acids between LRR14 and 15 [134]. Recognition of various PAMPs by the N-terminal domain causes TLR molecules to undergo conformational changes in the TIR domain that facilitate binding of various intracellular adaptor molecules with the TIR domain, thereby triggering a cascade signaling mechanism [99].
Initially, TLR9 is present in the endoplasmic reticulum (ER) [78], but after exposure of host cells to PAMPs, it migrates to endosomes [77]. In general, TLRs expressed in an endosomal membrane need to interact with a transmembrane protein of the ER, namely, Unc-93 homolog B1 (UNC-93B) as a prerequisite for endosomal translocation from the ER and subsequent recognition of PAMPs [22]. Such translocated TLRs, particularly TLR7 and TLR9, need further activation through cleavage of their N-terminal domain by endosomal proteases [31]. Activation of TLR9 is mediated by binding a stimulatory sequence of microbial DNA containing CpG motifs; however, a non-stimulatory sequence of microbial DNA can competitively block this activation [77, 110].
TLR signaling pathways
Activation of TLR signaling pathways results in maturation, differentiation and expansion of a number of immune cells, including macrophages, B cells, NK cells and T cells. Innate mediators activated downstream of TLR signaling coordinate recruitment of immune cells. In a recent study using a microarray to evaluate thousands of genes, CpG DNA treatment of host cells upregulated 77 genes, including IFNs, tumor necrosis factor-α (TNF-α), interleukin (IL)-6, IL-10, IL-12, cyclooxygenase-2 (COX-2), iNOS and granulocyte-macrophage colony-stimulating factor (GM-CSF) [62].
A TLR-ligand interaction initially stimulates conformational changes in the TIR domain, facilitating recruitment of various adaptor molecules, e.g., myeloid differentiation primary response-88 (MyD-88) protein, TIR domain-containing adaptor protein (TIRAP), and a TIR-domain-containing adaptor inducing IFN (TRIF) to initiate downstream signaling [52, 128]. Furthermore, MyD-88 is a key adaptor molecule involved in most of the activating signaling pathways of cell-surface and endosomal TLRs that are denoted MyD-88-dependent signaling pathways (Fig. 2) [88]. However, in studies with MyD-88 knockout mice/cell lines, NF-κB was not activated following TLR2, TLR7 and TLR9 stimulation [49, 63, 122]. However, ligands of TLR3 and TLR4 produce type 1 IFNs and delay activation of NF-κB, suggesting involvement of a MyD-88-independent signaling pathway [7, 30]. Furthermore, a TRIF protein is a key adaptor molecule for TLR3 activation pathway (designated as a TRIF-dependent signaling pathway or MyD-88-independent signaling pathway) [128]. In contrast, TLR4 may initiate both MyD-88-dependent and TRIF-dependent signaling pathways in mammals [152]. In chickens, TLR4 activation may upregulate TRIF mRNA expression [59], although an LPS-TLR4 interaction selectively activates only a MyD-88-dependent signaling pathway in chickens, and not both signaling pathways, as in mammals [65].
In the MyD-88-dependent signaling pathway, signaling initiated by TLR-associated MyD-88 molecules activates many cytoplasmic mediators, including IL-1 receptor-associated kinases (IRAKs) and TNF receptor-associated factor (TRAF) 6, resulting in activation of transforming growth factor beta-activated kinase-1 (TAK1). The latter protein ultimately has a dual role, activating either NF-κB or activator protein (AP) 1 through a series of reactions (Fig. 2) [145]. Thereafter, NF-κB or AP-1 enters the nucleus and upregulates gene transcription of innate and pro-inflammatory mediators including iNOS, IL-1, TNF-α and IL-6 (Fig. 2) [4, 145]. Furthermore, activation of the MyD-88-IRAK-TRAF6 complex by endosomal TLRs sequentially activates an additional pathway via activation of TRAF3, IRAK1 and IkappaB kinase-α (IKKα), ultimately leading to activation of IRF-7 [20, 141], which enters the nucleus and upregulates gene transcription for type 1 IFNs (potent antiviral cytokines) [142].
In the MyD-88-independant pathway (TRIF-dependent signaling pathway) [128], the activated TIR domain binds to TRIF (an adaptor molecule), which recruits receptor-interacting protein kinase 1 (RIP1) and TRAF6 molecules to activate TAK1, ultimately leading to activation of NF-κB, as in the MyD-88-dependent pathway [7, 147]. Alternatively, TRIF can activate the TRAF3-TANK-binding kinase 1 (TBK1)-IKKε complex, causing phosphorylation and activation of IRF3 and IRF7 [30, 118], which move into the nucleus and upregulate transcription for antiviral type-1 IFNs [118].
Cellular production and antiviral mechanisms of NO
NO production
It is well known that NO is a highly diffusible free radical molecule derived from L-arginine via NO synthase (NOS) enzyme activity in the presence of NADPH [95] and that it is widely involved in the regulation of various physiological mechanisms, including the immune, circulatory and nervous systems (Fig. 2). There are three isoforms of NOS: neuronal NOS (nNOS or NOS1), endothelial NOS (eNOS or NOS2) and inducible NOS (iNOS or NOS3) [143]. Both nNOS and eNOS are classified as constitutive NOS; they are less responsive to stimulation, have calmodulin/calcium-dependent enzyme systems [6], and produce low concentrations of NO [5]. Conversely, iNOS is mainly involved in the innate arm of the immune system, and its enzyme activity is calmodulin/calcium-independent [6, 87]. Furthermore, immunological stimulation (e.g., TLR signaling) is necessary for it to produce NO [87]. However, once stimulated, iNOS produces large quantities of NO for prolonged intervals, thereby facilitating innate host responses [75, 120].
Activation of TLR signalling pathways leading to NO production
Production of NO in host cells can be activated via various TLR pathways (Fig. 2). Of the cell-membrane-expressed TLRs, TLR2, 3, 4 and 5 are involved in signaling leading to NO production via activation of the MyD-88-dependent pathway [45]. Binding of LPS to TLR4 induces a strong NO response [45]. In a mouse macrophage cell line, LPS in combination with IFN-γ elicited production of much more NO than LPS alone [84], suggesting that mouse macrophages require priming with IFN-γ to enhance NO production. Similarly, in avian macrophages, priming with IFN-γ [23] or a viral infection [90] is a prerequisite for an LPS-mediated NO response. Gram-negative bacterial flagellin induces NO production in macrophages by a pathway involving both TLR4 and TLR5. Furthermore, TLR4 may promote binding of flagellin to TLR5/TLR4 complexes, as flagellin failed to induce NO production in cells with non-functional TLR4 [93]. The TLR2 ligand LTA induces iNOS expression and NO release from mouse macrophage cell line (RAW 264.7), and this is mediated via two pathways leading to NF-κB activation. An early response (minutes) is mediated by NF-κB activation via phosphatidylcholine-phospholipase activation, whereas in a late response (hours), NF-κB production is promoted by a COX2-prostaglandin E2-mediated pathway [19].
Of the endosomal-membrane-expressed TLRs, both TLR9/21 signaling and TLR3 signaling induce NO production. The presence of DNA containing CpG motifs, which serve as TLR9/21 ligands, induces NO production in macrophage cell lines; the stimulatory effect is positively correlated with a number of motifs in CpG DNA, e.g., GTCGTT [44]. Production of NO by avian macrophages was studied using various classes of CpG, viz. CpG 2216, CpG 2395, CpG 1826 and CpG 2007. Nearly all CpG classes (except CpG 2216) induced significant production of NO in comparison to non-CpG controls [8]. Furthermore, poly I:C, the TLR3 ligand, stimulated mouse bone marrow macrophages to produce NO [76], and it also activated iNOS in human monocyte-derived macrophages [125].
In addition to TLR signaling using single TLR ligands, additive effects of induction of multiple TLR pathways for NO production have been reported. For example, production of NO in avian PBMCs was higher with a combination of ligands than with only a single ligand, confirming previous observations [46]. Also LPS and microbial DNA had a synergistic effect in enhancing iNOS expression and production of NO in macrophages [36]. Similarly, a combination of recombinant flagellin and LPS synergistically induced NO production in avian PBMCs [40]. In addition, peptidoglycan (PepG) and LTA from Staphylococcus aureus synergistically induce iNOS. Finally, the level of NO production by murine macrophages in response to PepG, LTA and PepG plus LTA was much higher if two or more of these ligands were present [27].
Antiviral mechanisms of NO
A highly reactive free radical, NO has key roles in innate immune responses against numerous viruses [13, 24, 120] that infect mammals and birds [41, 70, 146]. It is well documented that induction of iNOS expression or providing NO by adding NO donors such as S-nitroso-N-acetylpenicillamine (SNAP) inhibits replication of various RNA or DNA viruses. Due to its hydrophobicity, NO diffuses freely across cell membranes (without receptors or carrier proteins) [21, 83]. An unpaired electron makes NO highly chemically reactive, and its antiviral mechanism is mainly a paracrine effect [73]. After diffusion through the cell membrane, NO may have several antiviral mechanisms. First, viruses with a DNA genome may undergo direct damage via nitrosation of primary amines [97, 131] in addition to indirect damage by endogenously produced NO-mediated N-nitrosamines, including N2O3 [92]. Second, NO indirectly reduces synthesis of viral genome and viral proteins in host cells by inactivating or modifying molecules involved in viral replication, including viral proteases [120], ribonucleotide reductase [80], reverse transcriptase [21, 104], transcriptional factors [123], and tyrosine- or heme-containing enzymes [54], through nitrosylation of these enzymes (Fig. 2) [130, 151].
This NO-mediated inhibition has been demonstrated in vitro against several viruses, including influenza virus [112, 113], dengue virus [129], herpes simplex virus [24, 89], vesicular stomatitis virus (VSV) [13], Japanese encephalitis virus [82], infectious laryngotracheitis virus [41], Marek’s disease virus (MDV) [146], coxsackievirus [34, 151], vaccinia virus [60], porcine respiratory coronavirus [57], rhinovirus [116], flavivirus [70], and hantavirus [67]. Similarly, based on in vivo studies, NO-mediated antiviral responses have been demonstrated against influenza virus [56], dengue virus [32], herpes simplex virus [10, 35], mouse hepatitis virus [74], Friend murine leukemia virus [3], hepatitis B virus [39], respiratory syncytial virus [126], infectious bursal disease virus [107], murine cytomegalovirus [98], MDV [146], coxsackievirus [34, 85], hantavirus [67], adenovirus [18] and VSV [69]. Antiviral responses observed in those in vitro and in vivo studies were based on exogenous NO supplied via NO donors. In addition to an antiviral effect, NO also has cytostatic effects in infected host cells, including inhibition of host DNA synthesis, protein synthesis and mitochondrial metabolism [105].
TLR activation leading to NO-mediated antiviral activity
Although there are numerous reports regarding NO-mediated antiviral responses, there is a paucity of literature on TLR-signaling-induced NO (endogenously produced)-mediated antiviral responses in mammals [3, 24, 50, 68, 85, 89] or birds [41, 105, 146]. A few in vitro studies have described NO-mediated antiviral activity following administration of TLR4 ligand against many viruses, including MDV, herpes simplex virus, infectious laryngotracheitis virus, coxsackievirus, and reovirus (Table 1). Similarly, an in vitro experiment with IRF3−/−IRF9−/− mouse embryo fibroblasts demonstrated NO-mediated antiviral activity using the TLR3 ligand poly I:C. In that study, iNOS inhibitors, including aminoguanidine hydrochloride (AMG) and N6-(1-iminoethyl)-L-lysine dihydrochloride (L-NIL), reversed the antiviral effect of poly I:C [89]. In an in vivo study in mice, coxsackievirus infection in combination with LPS (TLR4 ligand) induced expression of iNOS in macrophages. In addition, inhibition of NO production by NG-monomethyl-L-arginine (NMMA) increased viral load and mortality [85].
Viral constituents that activate the TLR pathways leading to NO-mediated antiviral responses are shown in Figure 3. In addition to viral nucleic acid, components of viral envelopes are also capable of inducing NO production, leading to antiviral activity. Induction of NO following viral replication in the host may curtail ongoing viral replication. However, prior induction of NO- mediated antiviral responses via induction of the TLR pathway appears to be more effective in preventing virus-induced pathology.

Viral components that activate TLRs signaling leading to NO-mediated antiviral activity in mammals and birds. In birds, TLR8 and TLR9 are absent; however, TLR21 replaces the function of TLR9 in mammals. TLR3, TLR7, TLR8, TLR9 and TLR21 are endosomal TLRs that recognize viral nucleic acids, whereas TLR2 and TLR4 are surface TLRs that detect viral surface molecules
NO and influenza virus
Although the role of NO against influenza virus infection is not well defined, in vitro, there are clearly beneficial effects of NO against influenza virus infections. Adding SNAP (an NO donor) to Madin-Darby canine kidney (MDCK) cells immediately after infection with influenza A and B viruses inhibited replication of both viruses in a dose-dependent manner during initial stages of infection [113]. Similarly, when the MDCK cell line was exposed to gaseous NO before and after infection with influenza A and B viruses, there was inactivation of viral neuraminidase activity and inhibition of viral infectivity by both pre- and post-infection NO exposures [112]. Unfortunately, studies conducted in vivo do not necessarily support observations made in in vitro systems. In a mouse model of influenza infection, inhalation of NO prior to influenza infection may decrease mouse survival, with no change in lung viral loads [26]. Using iNOS-knockout and wild-type mice, it was shown that NO production is not essential to clear infections with influenza virus A and reduce pulmonary pathology. In that experiment, wild-type mice that produced NO had severe pneumonitis [61], suggesting that NO was a detrimental factor exacerbating pneumonia during influenza virus infection [2]. Apparent discrepancies in observations between in vitro and in vivo studies regarding the benefits of NO against influenza virus infection in mammalian models may be explained by NO-mediated increases in pulmonary inflammation, which could be detrimental to the host in vivo [150]. Perhaps an appropriate balance between antiviral and inflammatory effects of NO is required for successful protective immunity against influenza virus infection in vivo [103].
Conclusions
Of the two TLR signaling pathways, the MyD-88-dependent signaling pathway is the one that predominantly leads to NO-mediated antiviral responses. Analysis of the current knowledge in the area of TLR-signaling-mediated NO-dependent antiviral responses revealed knowledge deficits in three major areas. Firstly, despite many reports regarding antiviral activity of NO against numerous mammalian and avian viruses based on the use of external NO donor compounds, there is a paucity of information on activation of TLR signaling pathways resulting in endogenous NO production leading to innate antiviral responses in mammalian and avian hosts. Secondly, of the studies that describe activation of TLR signaling pathways leading to NO-mediated antiviral responses, there are very few on NO-mediated antiviral responses in vivo [85]. Consequently, the therapeutic or prophylactic potential of activation of TLR signaling leading to NO-mediated antiviral responses is not well defined for either mammals or birds. Finally, although various MyD-88 signaling pathways are potentially capable of eliciting a NO response, most studies have focused on TLR4 and TLR3 signaling leading to NO-mediated antiviral responses, and further studies are therefore required to clarify the role of other MyD-88 signaling pathways in NO-mediated antiviral responses.
References
Abdul-Careem MF, Haq K, Shanmuganathan S, Read LR, Schat KA, Heidari M, Sharif S (2009) Induction of innate host responses in the lungs of chickens following infection with a very virulent strain of Marek’s disease virus. Virology 393:250–257
Akaike T, Noguchi Y, Ijiri S, Setoguchi K, Suga M, Zheng YM, Dietzschold B, Maeda H (1996) Pathogenesis of influenza virus-induced pneumonia: involvement of both nitric oxide and oxygen radicals. Proceed Nat Acad Sci USA 93:2448–2453
Akarid K, Sinet M, Desforges B, Gougerot-Pocidalo MA (1995) Inhibitory effect of nitric oxide on the replication of a murine retrovirus in vitro and in vivo. J Virol 69:7001–7005
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801
Aktan F (2004) iNOS-mediated nitric oxide production and its regulation. Life Sci 75:639–653
Alderton WK, Cooper CE, Knowles RG (2001) Nitric oxide synthases: structure, function and inhibition. Biochem J 357:593–615
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA (2001) Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738
Barjesteh N, Behboudi S, Brisbin JT, Villanueva AI, Nagy E, Sharif S (2014) TLR ligands induce antiviral responses in chicken macrophages. PloS One 9:e105713
Beklen A, Sorsa T, Konttinen YT (2009) Toll-like receptors 2 and 5 in human gingival epithelial cells co-operate with T-cell cytokine interleukin-17. Oral Microbiol Immunol 24:38–42
Benencia F, Courreges MC, Gamba G, Cavalieri H, Massouh EJ (2001) Effect of aminoguanidine, a nitric oxide synthase inhibitor, on ocular infection with herpes simplex virus in Balb/c mice. Invest Ophthalmol Visual Sci 42:1277–1284
Beutler B (2000) Tlr4: central component of the sole mammalian LPS sensor. Curr Opin Immunol 12:20–26
Beutler B, Jiang Z, Georgel P, Crozat K, Croker B, Rutschmann S, Du X, Hoebe K (2006) Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Ann Rev Immunol 24:353–389
Bi Z, Reiss CS (1995) Inhibition of vesicular stomatitis virus infection by nitric oxide. J Virol 69:2208–2213
Bieback K, Lien E, Klagge IM, Avota E, Schneider-Schaulies J, Duprex WP, Wagner H, Kirschning CJ, Ter Meulen V, Schneider-Schaulies S (2002) Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J Virol 76:8729–8736
Bittencourt VC, Figueiredo RT, da Silva RB, Mourao-Sa DS, Fernandez PL, Sassaki GL, Mulloy B, Bozza MT, Barreto-Bergter E (2006) An alpha-glucan of Pseudallescheria boydii is involved in fungal phagocytosis and Toll-like receptor activation. J Biol Chem 281:22614–22623
Brownlie R, Zhu J, Allan B, Mutwiri GK, Babiuk LA, Potter A, Griebel P (2009) Chicken TLR21 acts as a functional homologue to mammalian TLR9 in the recognition of CpG oligodeoxynucleotides. Mol Immunol 46:3163–3170
Brownlie R, Allan B (2011) Avian toll-like receptors. Cell Tissue Res 343:121–130
Cao W, Baniecki ML, McGrath WJ, Bao C, Deming CB, Rade JJ, Lowenstein CJ, Mangel WF (2003) Nitric oxide inhibits the adenovirus proteinase in vitro and viral infectivity in vivo. FASEB J Off Publ Feder Am Soc Experim Biol 17:2345–2346
Chang YC, Li PC, Chen BC, Chang MS, Wang JL, Chiu WT, Lin CH (2006) Lipoteichoic acid-induced nitric oxide synthase expression in RAW 264.7 macrophages is mediated by cyclooxygenase-2, prostaglandin E2, protein kinase A, p38 MAPK, and nuclear factor-kappaB pathways. Cell Signal 18:1235–1243
Chang ZL (2010) Important aspects of Toll-like receptors, ligands and their signaling pathways. Inflamm Res Off J Euro Histam Res Soc [et al] 59:791–808
Colasanti M, Persichini T, Venturini G, Ascenzi P (1999) S-nitrosylation of viral proteins: molecular bases for antiviral effect of nitric oxide. IUBMB Life 48:25–31
Conley ME (2007) Immunodeficiency: UNC-93B gets a toll call. Trends Immunol 28:99–101
Crippen TL, Sheffield CL, He H, Lowry VK, Kogut MH (2003) Differential nitric oxide production by chicken immune cells. Dev Comp Immunol 27:603–610
Croen KD (1993) Evidence for antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication. J Clin Invest 91:2446–2452
Dalpke AH, Heeg K (2004) CpG-DNA as immune response modifier. Int J Med Microb IJMM 294:345–354
Darwish I, Miller C, Kain KC, Liles WC (2012) Inhaled nitric oxide therapy fails to improve outcome in experimental severe influenza. Int J Med Sci 9:157–162
De Kimpe SJ, Kengatharan M, Thiemermann C, Vane JR (1995) The cell wall components peptidoglycan and lipoteichoic acid from Staphylococcus aureus act in synergy to cause shock and multiple organ failure. Proceed Nat Acad Sci USA 92:10359–10363
Dellacasagrande J (2009) Ligands, cell-based models, and readouts required for Toll-like receptor action. Methods Mol Biol 517:15–32
Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E, Szabo G (2004) Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 127:1513–1524
Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G (2002) IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 17:251–263
Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, Barton GM (2008) The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature 456:658–662
Fagundes CT, Costa VV, Cisalpino D, Amaral FA, Souza PR, Souza RS, Ryffel B, Vieira LQ, Silva TA, Atrasheuskaya A, Ignatyev G, Sousa LP, Souza DG, Teixeira MM (2011) IFN-gamma production depends on IL-12 and IL-18 combined action and mediates host resistance to dengue virus infection in a nitric oxide-dependent manner. PLoS Negl Tropical Dis 5:e1449
Feuillet V, Medjane S, Mondor I, Demaria O, Pagni PP, Galan JE, Flavell RA, Alexopoulou L (2006) Involvement of Toll-like receptor 5 in the recognition of flagellated bacteria. Proceed Nat Acad Sci USA 103:12487–12492
Flodstrom M, Horwitz MS, Maday A, Balakrishna D, Rodriguez E, Sarvetnick N (2001) A critical role for inducible nitric oxide synthase in host survival following coxsackievirus B4 infection. Virology 281:205–215
Gamba G, Cavalieri H, Courreges MC, Massouh EJ, Benencia F (2004) Early inhibition of nitric oxide production increases HSV-1 intranasal infection. J Med Virol 73:313–322
Gao JJ, Zuvanich EG, Xue Q, Horn DL, Silverstein R, Morrison DC (1999) Cutting edge: bacterial DNA and LPS act in synergy in inducing nitric oxide production in RAW 264.7 macrophages. J Immunol 163:4095–4099
Gay NJ, Gangloff M (2008) Structure of toll-like receptors. Handbook of experimental pharmacology:181–200
Gonzalez JM, Xu H, Ofori E, Elovitz MA (2007) Toll-like receptors in the uterus, cervix, and placenta: is pregnancy an immunosuppressed state? Am J Obst Gynecol 197(296):e291–e296
Guidotti LG, McClary H, Loudis JM, Chisari FV (2000) Nitric oxide inhibits hepatitis B virus replication in the livers of transgenic mice. J Experim Med 191:1247–1252
Gupta SK, Deb R, Gaikwad S, Saravanan R, Mohan CM, Dey S (2013) Recombinant flagellin and its cross-talk with lipopolysaccharide–effect on pooled chicken peripheral blood mononuclear cells. Res Veter Sci 95:930–935
Haddadi S, Kim DS, Jasmine H, van der Meer F, Czub M, Abdul-Careem MF (2013) Induction of Toll-like receptor 4 signaling in avian macrophages inhibits infectious laryngotracheitis virus replication in a nitric oxide dependent way. Veter Immunol Immunopathol 155:270–275
Hasan U, Chaffois C, Gaillard C, Saulnier V, Merck E, Tancredi S, Guiet C, Briere F, Vlach J, Lebecque S, Trinchieri G, Bates EE (2005) Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol 174:2942–2950
Haynes LM, Moore DD, Kurt-Jones EA, Finberg RW, Anderson LJ, Tripp RA (2001) Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J Virol 75:10730–10737
He H, Crippen TL, Farnell MB, Kogut MH (2003) Identification of CpG oligodeoxynucleotide motifs that stimulate nitric oxide and cytokine production in avian macrophage and peripheral blood mononuclear cells. Develop Comp Immunol 27:621–627
He H, Genovese KJ, Nisbet DJ, Kogut MH (2006) Profile of Toll-like receptor expressions and induction of nitric oxide synthesis by Toll-like receptor agonists in chicken monocytes. Mol Immunol 43:783–789
He H, Genovese KJ, Nisbet DJ, Kogut MH (2007) Synergy of CpG oligodeoxynucleotide and double-stranded RNA (poly I:C) on nitric oxide induction in chicken peripheral blood monocytes. Mol Immunol 44:3234–3242
Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S (2004) Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303:1526–1529
Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S (2000) A Toll-like receptor recognizes bacterial DNA. Nature 408:740–745
Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S (2002) Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol 3:196–200
Hiraoka Y, Kishimoto C, Takada H, Nakamura M, Kurokawa M, Ochiai H, Shiraki K (1996) Nitric oxide and murine coxsackievirus B3 myocarditis: aggravation of myocarditis by inhibition of nitric oxide synthase. J Am Coll Cardiol 28:1610–1615
Ho PP, Fontoura P, Ruiz PJ, Steinman L, Garren H (2003) An Immunomodulatory GpG Oligonucleotide for the Treatment of Autoimmunity via the Innate and Adaptive Immune Systems. J Immunol 171:4920–4926
Horng T, Barton GM, Medzhitov R (2001) TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol 2:835–841
Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G (2002) Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol 168:4531–4537
Ischiropoulos H (2003) Biological selectivity and functional aspects of protein tyrosine nitration. Biochem Biophy Res Commun 305:776–783
Jin MS, Lee JO (2008) Structures of the toll-like receptor family and its ligand complexes. Immunity 29:182–191
Jung K, Ha Y, Ha SK, Han DU, Kim DW, Moon WK, Chae C (2004) Antiviral effect of Saccharomyces cerevisiae beta-glucan to swine influenza virus by increased production of interferon-gamma and nitric oxide. J Veter Med B, Infect Dis Veter Publ Health 51:72–76
Jung K, Gurnani A, Renukaradhya GJ, Saif LJ (2010) Nitric oxide is elicited and inhibits viral replication in pigs infected with porcine respiratory coronavirus but not porcine reproductive and respiratory syndrome virus. Veter Immunol Immunopathol 136:335–339
Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, Liu YJ (2001) Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med 194:863–869
Karnati HK, Pasupuleti SR, Kandi R, Undi RB, Sahu I, Kannaki TR, Subbiah M, Gutti RK (2015) TLR-4 signalling pathway: MyD88 independent pathway up-regulation in chicken breeds upon LPS treatment. Veter Res Commun 39:73–78
Karupiah G, Harris N (1995) Inhibition of viral replication by nitric oxide and its reversal by ferrous sulfate and tricarboxylic acid cycle metabolites. J Exp Med 181:2171–2179
Karupiah G, Chen JH, Mahalingam S, Nathan CF, MacMicking JD (1998) Rapid interferon gamma-dependent clearance of influenza A virus and protection from consolidating pneumonitis in nitric oxide synthase 2-deficient mice. J Exp Med 188:1541–1546
Kato A, Homma T, Batchelor J, Hashimoto N, Imai S, Wakiguchi H, Saito H, Matsumoto K (2003) Interferon-alpha/beta receptor-mediated selective induction of a gene cluster by CpG oligodeoxynucleotide 2006. BMC Immunol 4:8
Kawai T, Adachi O, Ogawa T, Takeda K, Akira S (1999) Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11:115–122
Kawasaki T, Kawai T (2014) Toll-like receptor signaling pathways. Front Immunol 5:461
Keestra AM, van Putten JP (2008) Unique properties of the chicken TLR4/MD-2 complex: selective lipopolysaccharide activation of the MyD88-dependent pathway. J Immunol 181:4354–4362
Kielian T, Esen N, Bearden ED (2005) Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. Glia 49:567–576
Klingstrom J, Akerstrom S, Hardestam J, Stoltz M, Simon M, Falk KI, Mirazimi A, Rottenberg M, Lundkvist A (2006) Nitric oxide and peroxynitrite have different antiviral effects against hantavirus replication and free mature virions. Euro J Immunol 36:2649–2657
Koci MD, Kelley LA, Larsen D, Schultz-Cherry S (2004) Astrovirus-induced synthesis of nitric oxide contributes to virus control during infection. J Virol 78:1564–1574
Komatsu T, Bi Z, Reiss CS (1996) Interferon-gamma induced type I nitric oxide synthase activity inhibits viral replication in neurons. Journal of neuroimmunology 68:101–108
Kreil TR, Eibl MM (1996) Nitric oxide and viral infection: NO antiviral activity against a flavivirus in vitro, and evidence for contribution to pathogenesis in experimental infection in vivo. Virology 219:304–306
Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM (1995) CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 374:546–549
Krieg AM (2006) Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov 5:471–484
Lancaster JR Jr (1994) Simulation of the diffusion and reaction of endogenously produced nitric oxide. Proceed Nat Acad Sci USA 91:8137–8141
Lane TE, Paoletti AD, Buchmeier MJ (1997) Disassociation between the in vitro and in vivo effects of nitric oxide on a neurotropic murine coronavirus. J Virol 71:2202–2210
Laroux FS, Pavlick KP, Hines IN, Kawachi S, Harada H, Bharwani S, Hoffman JM, Grisham MB (2001) Role of nitric oxide in inflammation. Acta Physiol Scandin 173:113–118
Laszlo DJ, Henson PM, Remigio LK, Weinstein L, Sable C, Noble PW, Riches DW (1993) Development of functional diversity in mouse macrophages. Mutual exclusion of two phenotypic states. Am J Pathol 143:587–597
Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT (2004) TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol 5:190–198
Leifer CA, Kennedy MN, Mazzoni A, Lee C, Kruhlak MJ, Segal DM (2004) TLR9 is localized in the endoplasmic reticulum prior to stimulation. J Immunol 173:1179–1183
Leoni V, Gianni T, Salvioli S, Campadelli-Fiume G (2012) Herpes simplex virus glycoproteins gH/gL and gB bind Toll-like receptor 2, and soluble gH/gL is sufficient to activate NF-kappaB. J Virol 86:6555–6562
Lepoivre M, Fieschi F, Coves J, Thelander L, Fontecave M (1991) Inactivation of ribonucleotide reductase by nitric oxide. Biochem Biophy Res Commun 179:442–448
Lester SN, Li K (2014) Toll-like receptors in antiviral innate immunity. J Mol Biol 426:1246–1264
Lin YL, Huang YL, Ma SH, Yeh CT, Chiou SY, Chen LK, Liao CL (1997) Inhibition of Japanese encephalitis virus infection by nitric oxide: antiviral effect of nitric oxide on RNA virus replication. J Virol 71:5227–5235
Liu X, Srinivasan P, Collard E, Grajdeanu P, Zweier JL, Friedman A (2008) Nitric oxide diffusion rate is reduced in the aortic wall. Biophy J 94:1880–1889
Lowenstein CJ, Alley EW, Raval P, Snowman AM, Snyder SH, Russell SW, Murphy WJ (1993) Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proceed Nat Acad Sci USA 90:9730–9734
Lowenstein CJ, Hill SL, Lafond-Walker A, Wu J, Allen G, Landavere M, Rose NR, Herskowitz A (1996) Nitric oxide inhibits viral replication in murine myocarditis. J Clin Invest 97:1837–1843
Matsumoto M, Seya T (2008) TLR3: interferon induction by double-stranded RNA including poly(I:C). Adv Drug Deli Rev 60:805–812
Mayer B, Hemmens B (1997) Biosynthesis and action of nitric oxide in mammalian cells. Trends Biochem Sci 22:477–481
Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA Jr (1998) MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell 2:253–258
Mehta DR, Ashkar AA, Mossman KL (2012) The nitric oxide pathway provides innate antiviral protection in conjunction with the type I interferon pathway in fibroblasts. PloS One 7:e31688
Meyerhoff RR, Nighot PK, Ali RA, Blikslager AT, Koci MD (2012) Characterization of turkey inducible nitric oxide synthase and identification of its expression in the intestinal epithelium following astrovirus infection. Comp Immunol Microbiol Infect Dis 35:63–69
Michailidis G, Theodoridis A, Avdi M (2010) Transcriptional profiling of Toll-like receptors in chicken embryos and in the ovary during sexual maturation and in response to Salmonella enteritidis infection. Animal Reprod Sci 122:294–302
Miwa M, Stuehr DJ, Marletta MA, Wishnok JS, Tannenbaum SR (1987) Nitrosation of amines by stimulated macrophages. Carcinogenesis 8:955–958
Mizel SB, Honko AN, Moors MA, Smith PS, West AP (2003) Induction of macrophage nitric oxide production by Gram-negative flagellin involves signaling via heteromeric Toll-like receptor 5/Toll-like receptor 4 complexes. J Immunol 170:6217–6223
Mogensen TH, Paludan SR (2005) Reading the viral signature by Toll-like receptors and other pattern recognition receptors. J Mol Med (Berl) 83:180–192
Nathan C (1992) Nitric oxide as a secretory product of mammalian cells. FASEB J Off Publ Feder Am Soc Exp Biol 6:3051–3064
Nazli A, Kafka JK, Ferreira VH, Anipindi V, Mueller K, Osborne BJ, Dizzell S, Chauvin S, Mian MF, Ouellet M, Tremblay MJ, Mossman KL, Ashkar AA, Kovacs C, Bowdish DM, Snider DP, Kaul R, Kaushic C (2013) HIV-1 gp120 induces TLR2- and TLR4-mediated innate immune activation in human female genital epithelium. J Immunol 191:4246–4258
Nguyen T, Brunson D, Crespi CL, Penman BW, Wishnok JS, Tannenbaum SR (1992) DNA damage and mutation in human cells exposed to nitric oxide in vitro. Proceed Nat Acad Sci USA 89:3030–3034
Noda S, Tanaka K, Sawamura S, Sasaki M, Matsumoto T, Mikami K, Aiba Y, Hasegawa H, Kawabe N, Koga Y (2001) Role of nitric oxide synthase type 2 in acute infection with murine cytomegalovirus. J Immunol 166:3533–3541
O’Neill LA, Bowie AG (2007) The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nature Rev Immunol 7:353–364
O’Neill LA (2008) The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev 226:10–18
Ohto U, Shibata T, Tanji H, Ishida H, Krayukhina E, Uchiyama S, Miyake K, Shimizu T (2015) Structural basis of CpG and inhibitory DNA recognition by Toll-like receptor 9. Nature
Oosting M, Cheng SC, Bolscher JM, Vestering-Stenger R, Plantinga TS, Verschueren IC, Arts P, Garritsen A, van Eenennaam H, Sturm P, Kullberg BJ, Hoischen A, Adema GJ, van der Meer JW, Netea MG, Joosten LA (2014) Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proceed Nat Acad Sci USA 111:E4478–E4484
Pamer EG (2009) Tipping the balance in favor of protective immunity during influenza virus infection. Proceed Nat Acad Sci USA 106:4961–4962
Persichini T, Colasanti M, Fraziano M, Colizzi V, Medana C, Polticelli F, Venturini G, Ascenzi P (1999) Nitric oxide inhibits the HIV-1 reverse transcriptase activity. Biochem Biophy Res Commun 258:624–627
Pertile TL, Karaca K, Sharma JM, Walser MM (1996) An antiviral effect of nitric oxide: inhibition of reovirus replication. Avian Dis 40:342–348
Philbin VJ, Iqbal M, Boyd Y, Goodchild MJ, Beal RK, Bumstead N, Young J, Smith AL (2005) Identification and characterization of a functional, alternatively spliced Toll-like receptor 7 (TLR7) and genomic disruption of TLR8 in chickens. Immunology 114:507–521
Poonia B, Charan S (2005) Early and transient induction of nitric oxide (NO) in infectious bursal disease virus infection is T-cell dependent: a study in cyclosporin-A treated chicken-model. Indian J Exp Biol 43:192–196
Raetz M, Kibardin A, Sturge CR, Pifer R, Li H, Burstein E, Ozato K, Larin S, Yarovinsky F (2013) Cooperation of TLR12 and TLR11 in the IRF8-dependent IL-12 response to Toxoplasma gondii profilin. J Immunol 191:4818–4827
Rallabhandi P, Bell J, Boukhvalova MS, Medvedev A, Lorenz E, Arditi M, Hemming VG, Blanco JC, Segal DM, Vogel SN (2006) Analysis of TLR4 polymorphic variants: new insights into TLR4/MD-2/CD14 stoichiometry, structure, and signaling. J Immunol 177:322–332
Rankin R, Pontarollo R, Ioannou X, Krieg AM, Hecker R, Babiuk LA, van Drunen Littel-van den Hurk S (2001) CpG motif identification for veterinary and laboratory species demonstrates that sequence recognition is highly conserved. Anti Nucl Acid Drug Dev 11:333–340
Rassa JC, Meyers JL, Zhang Y, Kudaravalli R, Ross SR (2002) Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proceed Nat Acad Sci USA 99:2281–2286
Regev-Shoshani G, Vimalanathan S, McMullin B, Road J, Av-Gay Y, Miller C (2013) Gaseous nitric oxide reduces influenza infectivity in vitro. Nitric Oxide Biol Chem Off J Nitr Oxide Soc 31:48–53
Rimmelzwaan GF, Baars MM, de Lijster P, Fouchier RA, Osterhaus AD (1999) Inhibition of influenza virus replication by nitric oxide. J Virol 73:8880–8883
Rolland A, Jouvin-Marche E, Viret C, Faure M, Perron H, Marche PN (2006) The envelope protein of a human endogenous retrovirus-W family activates innate immunity through CD14/TLR4 and promotes Th1-like responses. J Immunol 176:7636–7644
Salazar Gonzalez RM, Shehata H, O’Connell MJ, Yang Y, Moreno-Fernandez ME, Chougnet CA, Aliberti J (2014) Toxoplasma gondii- derived profilin triggers human toll-like receptor 5-dependent cytokine production. J Innate Immun 6:685–694
Sanders SP, Siekierski ES, Porter JD, Richards SM, Proud D (1998) Nitric oxide inhibits rhinovirus-induced cytokine production and viral replication in a human respiratory epithelial cell line. J Virol 72:934–942
Sasai M, Yamamoto M (2013) Pathogen recognition receptors: ligands and signaling pathways by Toll-like receptors. Int Rev Immunol 32:116–133
Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T (2000) Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13:539–548
Sato M, Sano H, Iwaki D, Kudo K, Konishi M, Takahashi H, Takahashi T, Imaizumi H, Asai Y, Kuroki Y (2003) Direct binding of Toll-like receptor 2 to zymosan, and zymosan-induced NF-kappa B activation and TNF-alpha secretion are down-regulated by lung collectin surfactant protein A. J Immunol 171:417–425
Saura M, Zaragoza C, McMillan A, Quick RA, Hohenadl C, Lowenstein JM, Lowenstein CJ (1999) An antiviral mechanism of nitric oxide: inhibition of a viral protease. Immunity 10:21–28
Schenk M, Belisle JT, Modlin RL (2009) TLR2 looks at lipoproteins. Immunity 31:847–849
Schnare M, Holt AC, Takeda K, Akira S, Medzhitov R (2000) Recognition of CpG DNA is mediated by signaling pathways dependent on the adaptor protein MyD88. Curr Biol CB 10:1139–1142
Sekkai D, Aillet F, Israel N, Lepoivre M (1998) Inhibition of NF-kappaB and HIV-1 long terminal repeat transcriptional activation by inducible nitric oxide synthase 2 activity. J Biol Chem 273:3895–3900
Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M (1999) MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. The Journal of experimental medicine 189:1777–1782
Snell JC, Chernyshev O, Gilbert DL, Colton CA (1997) Polyribonucleotides induce nitric oxide production by human monocyte-derived macrophages. J Leukocyte Biol 62:369–373
Stark JM, Khan AM, Chiappetta CL, Xue H, Alcorn JL, Colasurdo GN (2005) Immune and functional role of nitric oxide in a mouse model of respiratory syncytial virus infection. J Infect Dis 191:387–395
Tada H, Nemoto E, Shimauchi H, Watanabe T, Mikami T, Matsumoto T, Ohno N, Tamura H, Shibata K, Akashi S, Miyake K, Sugawara S, Takada H (2002) Saccharomyces cerevisiae- and Candida albicans-derived mannan induced production of tumor necrosis factor alpha by human monocytes in a CD14- and Toll-like receptor 4-dependent manner. Microbiol Immunol 46:503–512
Takeda K, Akira S (2004) TLR signaling pathways. Sem Immunol 16:3–9
Takhampunya R, Padmanabhan R, Ubol S (2006) Antiviral action of nitric oxide on dengue virus type 2 replication. J General Virol 87:3003–3011
Tamir S, Burney S, Tannenbaum SR (1996) DNA damage by nitric oxide. Chem Res Toxicol 9:821–827
Tamir S, deRojas-Walker T, Wishnok JS, Tannenbaum SR (1996) DNA damage and genotoxicity by nitric oxide. Methods Enzymol 269:230–243
Tangeras LH, Stodle GS, Olsen GD, Leknes AH, Gundersen AS, Skei B, Vikdal AJ, Ryan L, Steinkjer B, Myklebost MF, Langaas M, Austgulen R, Iversen AC (2013) PP042. Cell surface toll-like receptors in primary first trimester trophoblasts. Pregnan Hyperten 3:81–82
Tanimura N, Saitoh S, Matsumoto F, Akashi-Takamura S, Miyake K (2008) Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem Biophy Res Commun 368:94–99
Tanji H, Ohto U, Shibata T, Miyake K, Shimizu T (2013) Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 339:1426–1429
Tapping RI (2009) Innate immune sensing and activation of cell surface Toll-like receptors. Semin Immunol 21:175–184
Temperley ND, Berlin S, Paton IR, Griffin DK, Burt DW (2008) Evolution of the chicken Toll-like receptor gene family: a story of gene gain and gene loss. BMC Genom 9:62
Thapa S, Cader MS, Murugananthan K, Nagy E, Sharif S, Czub M, Abdul-Careem MF (2015) In ovo delivery of CpG DNA reduces avian infectious laryngotracheitis virus induced mortality and morbidity. Viruses 7:1832–1852
Thapa S, Nagy E, Abdul-Careem MF (2015) In ovo delivery of Toll-like receptor 2 ligand, lipoteichoic acid induces pro-inflammatory mediators reducing post-hatch infectious laryngotracheitis virus infection. Veter Immunol Immunopathol 164:170–178
Thomas PG, Carter MR, Atochina O, Da’Dara AA, Piskorska D, McGuire E, Harn DA (2003) Maturation of dendritic cell 2 phenotype by a helminth glycan uses a Toll-like receptor 4-dependent mechanism. J Immunol 171:5837–5841
Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA (2011) Pattern recognition receptors and the innate immune response to viral infection. Viruses 3:920–940
Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M (2010) Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nature Immunol 11:70–75
Uematsu S, Akira S (2007) Toll-like receptors and Type I interferons. The Journal of biological chemistry 282:15319–15323
Villanueva C, Giulivi C (2010) Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radical Biol Med 49:307–316
Vollmer J, Krieg AM (2009) Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv Drug Del Rev 61:195–204
West AP, Koblansky AA, Ghosh S (2006) Recognition and signaling by toll-like receptors. Ann Rev Cell Dev Biol 22:409–437
Xing Z, Schat KA (2000) Inhibitory effects of nitric oxide and gamma interferon on in vitro and in vivo replication of Marek’s disease virus. J Virol 74:3605–3612
Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S (2003) Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301:640–643
Yilmaz A, Shen S, Adelson DL, Xavier S, Zhu JJ (2005) Identification and sequence analysis of chicken Toll-like receptors. Immunogenetics 56:743–753
Zahringer U, Lindner B, Inamura S, Heine H, Alexander C (2008) TLR2 - promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology 213:205–224
Zaki MH, Okamoto, T., Sawa, T., Fujii, S. and Akaike, T. (2007) Nitrative stress in respiratory inflammation caused by influenza virus infection. . Clin Exp Allergy Rev:8
Zell R, Markgraf R, Schmidtke M, Gorlach M, Stelzner A, Henke A, Sigusch HH, Gluck B (2004) Nitric oxide donors inhibit the coxsackievirus B3 proteinases 2A and 3C in vitro, virus production in cells, and signs of myocarditis in virus-infected mice. Med Microbiol Immunol 193:91–100
Zughaier SM, Zimmer SM, Datta A, Carlson RW, Stephens DS (2005) Differential induction of the toll-like receptor 4-MyD88-dependent and -independent signaling pathways by endotoxins. Infect Immun 73:2940–2950
Acknowledgements
The graduate programs of M.S.A and A.A. are supported by the Margaret Gunn Endowment for Animal Research (University of Calgary), Agriculture and Agri-Food Canada, and Saskatchewan Agriculture Ministry (Canada) grants received by MFA. We thank Dr. John Kastelic, University of Calgary Faculty of Veterinary Medicine, for extensive editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Margaret Gunn Endowment for Animal Research, University of Calgary, Canada (10007898); Agriculture and Agri-Food Canada (10009962); Saskatchewan Agriculture Ministry, Canada (10010719).
Conflict of interest
Mohamed Sarjoon Abdul-Cader declares that he has no conflict of interest. Aruna Amarasinghe declares that he has no conflict of interest. Mohamed Faizal Abdul-Careem declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Abdul-Cader, M.S., Amarasinghe, A. & Abdul-Careem, M.F. Activation of toll-like receptor signaling pathways leading to nitric oxide-mediated antiviral responses. Arch Virol 161, 2075–2086 (2016). https://doi.org/10.1007/s00705-016-2904-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-2904-x