
Abstract
Malignant epithelial tumors (carcinomas) are the most common ovarian cancers and also the most lethal gynecological malignancies. Based on histopathology and molecular genetic alterations, ovarian carcinomas are divided into five main types (high-grade serous (70%), endometrioid (10%), clear cell (10%), mucinous (3%), and low-grade serous carcinomas (<5%)) that account for over 95% of cases. These types are essentially distinct diseases, as indicated by differences in epidemiological and genetic risk factors, precursor lesions, patterns of spread, and molecular events during oncogenesis, response to chemotherapy, and prognosis. For a successful specific treatment, reproducible histopathological diagnosis of the tumor cell type is critical. The five tumor types are morphologically diverse and resemble carcinomas of the uterus. Actually, recent investigations have demonstrated that a significant number of cancers, traditionally thought to be primary ovarian tumors (particularly serous, endometrioid, and clear cell carcinomas), originate in the fallopian tube and the endometrium and involve the ovary secondarily. This review summarizes recent advances in the molecular pathology which have greatly improved our understanding of the biology of ovarian carcinoma and are also relevant to patient management.
Similar content being viewed by others


Epithelial ovarian tumors are heterogeneous neoplasms which are primarily classified according to cell type into serous, mucinous, endometrioid, clear cell, transitional, and squamous cell tumors [1, 2]. Parenthetically, none of these cells are found in the normal ovary and their development has long been attributed to mullerian “neometaplasia” of the ovarian surface epithelium (mesothelium). More importantly, these tumors are further subdivided into benign, borderline (intermediate), and malignant (carcinoma) depending upon the degree of cell proliferation and nuclear atypia, and the presence or absence of stromal invasion [1, 2].
Borderline tumors show epithelial proliferation greater than that seen in their benign counterparts and variable nuclear atypia; however, in contrast to carcinomas, there is absence of stromal invasion, and their prognosis is much better than that of carcinomas. In spite of the lack of ovarian stromal invasion, serous borderline tumors, particularly those with exophytic growth, can implant on peritoneal surfaces and, rarely (about 10% of peritoneal implants), progress to low-grade serous carcinoma (LGSC) and invade the underlying tissues. The biologic behavior of invasive peritoneal implants is similar to that of LGSC. However, the lack of stromal invasion in the primary borderline ovarian tumor justifies designating the associated (frequently superficial) peritoneal lesions as implants rather than metastasis [1, 2]. In contrast, LGSC is usually associated with peritoneal carcinomatosis [3].
Malignant epithelial tumors (carcinomas) are the most common ovarian cancers accounting for 90% of cases [1, 2]. Although traditionally referred to as a single entity, ovarian cancer is not a homogeneous disease but rather a group of diseases, each with different morphology and biologic behavior. Compared to breast cancer, ovarian cancer is ten times less frequent, yet is associated with a much greater number of deaths, as 75% of patients present with advanced (stage III) tumors experience recurrence after surgery and chemotherapy and most, ultimately, die of the disease. Globally, it accounts for over 100,000 women’s deaths per year, constitutes the fifth most frequent cause of cancer death in women in the Western world, and is the most lethal gynecological cancer [4]. Early diagnosis has been unsuccessful.
Unlike colorectal carcinoma, a progression model for ovarian carcinoma has not been described. In spite of being an oversimplified model, defining colorectal cancer as a linear sequence of mutations has served as a working guide and has allowed for a better understanding of tumor progression from premalignant lesions to invasive carcinoma [5]. The clinical benefit of this includes the successful screening, early detection, and treatment of colon cancer. Conversely, the origins of ovarian cancer are only now being elucidated, and thus it remains the most aggressive gynecologic malignancy.
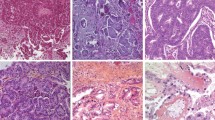
Currently, however, based on histopathology, immunohistochemistry, and molecular genetic analysis, at least five main types of ovarian carcinomas are identified: high-grade serous carcinomas (HGSC) (70%), endometrioid carcinomas (EC) (10%), clear cell carcinomas (CCC) (10%), mucinous carcinomas (MC) (3 %), and LGSC (<5%) [6] (Table 1) (Fig. 1). These tumors account for 98% of ovarian carcinomas, can be reproducibly diagnosed by light microscopy, and are inherently different diseases, as indicated by differences in epidemiological and genetic risk factors, precursor lesions, patterns of spread, molecular events during oncogenesis, response to chemotherapy, and prognosis.

Representative examples of the five main types of ovarian carcinoma, which together account for 98% of cases: a High-grade serous carcinoma; b Low-grade serous carcinoma; c Mucinous carcinoma; d Endometrioid carcinoma; and e Clear cell carcinoma
In the era of personalized cancer medicine, reproducible histopathological diagnosis of tumor cell type is a sine qua non condition for successful treatment. For instance, it has been found that different tumor types respond differently to chemotherapy. The poor response rate of clear cell carcinomas (15%) contrasts notably with that of high-grade serous carcinomas (80%), resulting in a lower 5-year survival for clear cell compared with high-grade serous carcinoma in patients with advanced stage tumors (20% versus 30%) [7, 8]. The clear cell and mucinous types, in particular, are candidates for clinical trials to identify more active therapy than what is presently used [9]. Furthermore, the biomarker expression profile within a given type is consistent across stage [10]. Thus, early and advanced stage ovarian carcinomas differ primarily based on histological type, while, within a type there is no difference in biomarker expression between early and advanced stage tumors [9].
The fact that one tumor type (high-grade serous carcinomas) accounts for over two-thirds of cases, it does not justify classifying ovarian carcinomas into only two types, lumping together the other four (endometrioid, clear cell, mucinous, and low-grade serous carcinomas) as “type 1 carcinomas” [11]. In fact, the latter tumors are clinically, morphologically, and molecularly distinct diseases that individually bear resemblance neither to high-grade serous carcinomas nor to each other. Thus, classifying ovarian carcinomas into just two types (“types I and II”) [11] is artificial and limits progress in understanding the biology or improving the management of the less common types of ovarian carcinomas.
As indicated above, benign counterparts of the cells of ovarian carcinomas are not found in the normal ovary. The tumor cells were thought to derive exclusively from the surface epithelium (mesothelium) and epithelial inclusion cysts through a process of mullerian “neometaplasia”; thus, they would resemble morphologically the epithelia of the fallopian tube, endometrium, or endocervix. Although the mesothelial origin cannot be excluded, there is now compelling evidence that a number of what have been thought to be primary ovarian cancers are actually originated in other pelvic organs and involve the ovary secondarily. In fact, it has been proposed that HGSC arise from precursor epithelial lesions in the distal fimbriated end of the fallopian tube [12–18], whereas endometrioid and clear cell carcinomas originate from ovarian endometriosis (Fig. 2) [19, 20]. This review summarizes recent advances in the molecular pathology which have greatly improved our understanding of the biology of ovarian carcinoma and are also relevant to patient management.

Classification of gynecological cancers based on origin and mutations
Serous carcinomas
It is now accepted that high-grade serous carcinoma and low-grade serous carcinoma are fundamentally different tumor types, and consequently, different diseases [20]. LGSCs are associated, in most cases, with a serous borderline component, carry KRAS and BRAF mutations, and are unrelated to TP53 mutations and BRCA abnormalities [21, 22]. In contrast, HGSCs are not associated with serous borderline tumors and typically exhibit TP53 mutations and BRCA abnormalities. However, it has been recently shown that both tumor types may derive from fallopian tube precursor lesions (tubal intraepithelial carcinoma (TIC)) [12–18] (Fig. 3) or fallopian tube epithelium (endosalpingiosis) in a significant number of cases [23, 24].

Tubal intraepithelial carcinoma (a). Immunostaining for p53 and Ki-67 are shown as (b) and (c), respectively. Note the abrupt transition from benign tubal epithelium to a region with marked cytological atypia, diffuse strong p53 immunoreactivity, and increased proliferation index
High-grade serous carcinomas
HGSCs are the most common ovarian carcinomas and most patients present with advanced stage disease (approximately 80%); tumors confined to the ovary at diagnosis are distinctly uncommon (<10%).
Microscopically, HGSCs show papillary and solid growth with slit-like glandular lumens. The tumor cells are typically of intermediate size, with scattered bizarre mononuclear giant cells exhibiting prominent nucleoli (Fig. 1a). In contrast to LGSCs, these tumors show more than 3-fold variation in nuclear size. Although nuclear features are the chief criterion for distinguishing between HGSC and LGSC, the mitotic activity can be used in cases with equivocal degrees of nuclear pleomorphism; mitotic activity greater than 12/10 high-power microscopic fields (HPF) favors a diagnosis of HGSC [6, 25]. In these tumors, mitotic activity is often several times higher than the diagnostic threshold and is associated with abundant apoptotic bodies. High-grade and predominantly solid carcinomas showing serous differentiation, even in a minority of the tumor, should be classified as HGSC (rather than mixed serous/undifferentiated). To date, no underlying molecular differences between these tumors and pure HGSC have been detected [26].
Most HGSCs immunoreact for p53, BRCA1, WT1, and p16. They also exhibit a high proliferation index as indicated by an increased nuclear expression of Ki-67. Only strong and diffuse p53 and p16 reactions should be considered positive. Nuclear WT1 reaction occurs in approximately 80% of cases of HGSC and LGSC but in less than 5% of ovarian carcinomas of other types [10, 27, 28]. Estrogen receptor (ER) is expressed in approximately two thirds of cases of HGSC and is also expressed in LGSCs and ECs but is negative in almost all CCCs and MCs [28].
The traditional view that HGSC arise exclusively from ovarian surface epithelium or epithelial inclusion cysts has been recently challenged by the identification, in women with BRCA1 or BRCA2 germline mutations, of TIC in the distal fimbriated end of the fallopian tube as the probable precursor of advanced HGSC [12, 13, 16]. Cytologically, the cells of TIC show secretory differentiation [16] and resemble those of HGSC (Fig. 3a). They lack cilia and show nuclear enlargement, hyperchromasia, loss of polarity, prominent nucleoli, and mitotic figures. TIC shows strong immunoreaction for HMFG2, an antibody that recognizes mucin 1, and Stathmin 1, both markers of secretory cells, suggesting that these cells are the target for malignant transformation [16, 29]. TIC also shows immunohistochemical evidence of double-stranded DNA damage, as indicated by nuclear staining for gamma-H2AX [18]. Like HGSC, TIC diffusely and strongly expresses p53 (Fig. 3b) and the Ki-67 proliferation index is usually markedly elevated (Fig. 3c) (mean labeling index, 72%; range: 40–95%) [18]. p16 and WT-1 may also be expressed. Furthermore, the finding of identical TP53 mutations in both TIC and concomitant tumors classified as ovarian in origin [15] indicates a clonal relationship between them and suggests that the distal fallopian tube (fimbria) is an important site for the initiation of HGSC. Nevertheless, implantation of tubal-type epithelium into the ovary (endosalpingiosis) or mesothelial surface invaginations (inclusion cysts) may explain the origin of those HGSC lacking TIC. In such cases, the primary tumor would appear to originate from the ovary. Currently, the relative proportion of HGSC of ovarian and tubal derivation is unknown mainly because the growth of tumor in advanced stage cancers conceals the primary site. However, extensive examination of the fallopian tubes from 55 consecutive cases of HGSC (ovarian, tubal, or pelvic) revealed involvement of the endosalpinx in 70% and TIC in approximately 50% of the cases [15].
Women with germline mutations in BRCA1 or BRCA2 have a 30%–70% risk of developing ovarian cancer by the age of 70, mainly HGSC [30]. Carcinomas arising in patients with germline BRCA1 or BRCA2 mutations are almost invariably of high-grade serous type. BRCA1 and BRCA2 are essential components of the homologous recombination DNA system required to repair DNA double-strand breaks (DSB) [31]. Like TP53 mutations, BRCA inactivation seems to be a consistent genetic alteration of HGSC. Besides germline mutation, inactivation of the BRCA pathway may result from somatic mutation in either BRCA1 or BRCA2 [32], or promoter hypermethylation in BRCA1 [33].
The discovery of TICs in risk-reducing salpingo-oophorectomy (RRSO) specimens from women with known BRCA mutations and/or a strong family history of ovarian cancer has resulted in extensive research into the role of the fallopian tube in pelvic serous carcinogenesis [12–18]. Early studies revealed small foci of strongly p53-immunoreactive cells in largely histologically normal fallopian tube epithelium [16]. These foci, which predominate in the distal portion of the fallopian tube, have been designated “p53 signatures”. Like TIC, p53 signatures are comprised exclusively of secretory cells (at least 12 consecutive immunoreactive cells), and the majority exhibit evidence of DNA damage by immunoreaction for gamma-H2AX [16, 18]. They are more frequent and multifocal in tubes with TIC and, in some cases, can be identified in direct continuity with TIC. About 57% of p53 signatures contain TP53 mutations [16]; however, Ki-67 proliferation index is low (mean, 3%). p53 signatures probably represent early clonal expansion short of neoplastic proliferation [34] and, surprisingly, are found in both women with and without BRCA1 or BRCA2 mutations at the same frequency (10–38% versus 17–33%, respectively) [16]; should BRCA loss have caused p53 signature foci, one would expect a much higher frequency in women with germline mutations in BRCA1 or BRCA2, but this does not occur [16]. Thus, TP53 mutation is an early event in the genesis of HGSC, occurring in p53 signature foci and leading to TIC in the distal fallopian tube. BRCA1 mutation also occurs early in the development of TIC but after TP53 mutation [34]. It is possible that germline mutations of BRCA1 act as a promoter for the development of TIC [17].
Jarboe et al. have described a morphologic continuum of epithelial changes taking place in the distal fallopian tube [18]. The transition is as follows: normal fallopian tube epithelium, overexpression of p53, serous tubal intraepithelial carcinoma (STIC), and finally, invasive serous carcinoma. Clonality of the precursor cells in both the so-called p53 signature and STICs are the strongest support for the fimbriated end of the fallopian tube as a site of origin for HGSC [14]. Concentration of TP53 mutant lesions in the distal fallopian tube suggests a vulnerability of these cells to DNA damage. In fact, the secretory cells of the tubal epithelium have a limited ability to repair DNA DSB, as shown by the persistence of gamma-H2AX immunoreactive foci after DNA damage [35]. This might explain why this tissue seems to be especially sensitive to inactivating BRCA mutations.
The protocol for Sectioning and Extensively Examining the Fimbriated End (SEE-FIM protocol) was developed for processing RRSO [14]. According to this protocol, the entire tube is initially fixed for at least 4 h to prevent denuding of the mucosal epithelial cells. Then, the fimbriated end is amputated from the proximal tube and sectioned longitudinally into multiple (at least four) sections and the entire tube is submitted for histologic review. With more extensive sectioning of these specimens, an increased rate of detection of early cancer (up to 17%) can be obtained [36].
Although a significant number of HGSC may not arise from the ovary, and the term “ovarian cancer” would not be pathogenetically precise in every case, ovarian involvement is the rule in almost all cases. Furthermore, in view of the rarity of HGSC associated with tubal tumor masses, it is unlikely that all HGSC originate in the fallopian tube. Thus, the term HGSC of ovary should be kept until the different origins of the ovarian tumors are better understood. Terms like “mullerian” or “pelvic” would create confusion for patients, physicians, and medical investigators.
A pathogenetic model that includes the stages of initiation and progression of HGSC is essential for an effective screening and treatment that takes into account biomarkers of early tumorigenesis. The model described by Bowtell [34] (Fig. 4) proposes that the sequence of primary events were as follows: early p53 loss followed by BRCA loss, leading to deficiency in homologous recombination repair of DSB, which triggers chromosomal instability (genetic chaos) and widespread copy number changes [34, 37, 38]. Secondary and tertiary events then cause global changes in gene expression followed by mutations to facilitate tumor evolution. Importantly, this model suggests early loss of p53 and BRCA. Once chromosomal instability is set up by mutation in TP53 and BRCA inactivation, gene copy number is the major determinant of progression of HGSC [34]. Potential biomarkers or therapeutic targets must consider the driver mutations and events that occur before the full development of carcinoma, when a multitude of mutations and copy number alterations occur to configure each individual tumor.

Initiation and progression of high-grade serous ovarian cancer. This model proposes that the sequence of primary events were as follows: early p53 loss followed by BRCA loss, leading to deficiency in homologous recombination repair (HRR) of DSB, which triggers chromosomal instability (genetic chaos) and widespread copy number changes. Copy number change can be a driver of molecular subtype specification and results in global changes in gene expression. Subsequent mutations facilitate tumor progression. Modified from DD Bowtell [34]
Concerning heterogeneity of HGSC, we have recently described two molecular subtypes of this tumor based on distinct patterns of expression of genes involved in the PI3K–AKT pathway [39]. Expression of active caspase-3 in the tumor stroma (lymphocytes and macrophages) was associated with good prognosis and response to chemotherapy. Furthermore, co-expression of caspase-3 and X-linked inhibitor of apoptosis (XIAP) identified high-grade serous carcinomas with better prognosis. These findings indicate that there are different biological subtypes of HGSC.
After primary surgical debulking, most (70–80%) HGSCs show a response to platinum/taxane chemotherapy, but the majority of patients will subsequently experience a recurrence, at which point cure is not possible [8]. Recently, PARP inhibitors that target loss of BRCA function and inability to repair double-strand breaks in DNA have been used [40, 41].
Low-grade serous carcinomas
LGSCs are uncommon and account for less than 5% of all cases of ovarian carcinoma [42]. They frequently have a noninvasive serous borderline component (with or without micropapillary pattern) and most likely represent progression of serous borderline tumors beyond microinvasion. Whereas the presence of small foci of LGSC in an ovarian borderline tumor is associated with an excellent prognosis, patients with advanced stage disease fare less favorably. Nevertheless, the disease usually follows a relatively indolent course.
Microscopically, LGSCs show small papillae of tumor cells exhibiting uniform nuclei within variable amounts of hyalinized stroma, which often contains psammoma bodies (Fig. 1b). Uniformity of the nuclei is the principal criterion for distinguishing between LGSC and HGSC, with less than threefold variability [25]. This distinction has been shown to be highly reproducible [43]. Tumors showing nuclei of intermediate size often have TP53 mutations and should be classified as HGSC [44]. LGSC rarely progress to high-grade tumors.
The biomarker expression profile of LGSC is similar to that of their high-grade counterparts. Only Ki-67 immunoreaction differs significantly between the two tumor types, with median Ki-67 labeling index of 2.5% in LGSC versus 22.4% in HGSC [10]. BRAF or KRAS mutations are present in LGSCs (38% and 19%, respectively). [21, 45]. LGSCs do not show chromosomal instability and lack the complex genetic abnormalities seen in HGSCs. LGSCs are not associated with BRCA germline mutations.
With regard to the distinction between LGSC and serous borderline tumor, micropapillarity, by itself, is not sufficient to warrant a diagnosis of carcinoma in the absence of invasion. If there are invasive foci measuring less than 10 mm2, the tumor is considered to be borderline with microinvasion [46]. Tumors with larger invasive components are classified as LGSC. Histopathologically, invasive peritoneal implants and LGSC are identical lesions which are only distinguished by the timing of the disease and the volume of the tumor. Whereas invasive implants are early superficial lesions of microscopic or small macroscopic size (≤1–2 cm), LGSC frequently presents as bulky disease [3]. Although the independent origin of the invasive peritoneal implants associated with ovarian SBT cannot be completely excluded, we have recently demonstrated identical BRAF and KRAS mutations as well as identical LOH in a series of ovarian SBT and peritoneal implants. Such findings support a monoclonal origin of these tumors and the secondary nature of the implants [47].
The response rate to conventional therapy for LGSC is difficult to determine because this tumor type has only recently been recognized and existing data may reflect case series that include some cases of HGSC. Data from series of patients with serous borderline tumors who experience a recurrence as carcinoma, indicate that, in most cases, LGSC do not respond to conventional ovarian carcinoma chemotherapy [48].
Mucinous carcinomas
Mucinous tumors account for 10–15% of all primary ovarian tumors; however, approximately 80% are benign and most of the remainder are borderline tumors. If metastases to the ovary, particularly from the gastrointestinal tract, are carefully excluded, only 3–4% of ovarian carcinomas are of mucinous type. The cells of MCs may resemble those of the gastric pylorus, intestine, or endocervix [1, 2]; nevertheless, the vast majority of these tumors show gastrointestinal differentiation (Fig. 1c). The origin of these tumors is unknown. Large size (>13 cm) and unilaterality are features suggestive of a primary MC, while metastases are typically smaller and bilateral. Primary MCs of the ovary are usually confined to the ovary, without ovarian surface involvement or pseudomyxoma peritonei.
Malignant mucinous ovarian tumors are often heterogeneous. Benign-appearing, borderline, noninvasive carcinoma, and invasive components may coexist within an individual tumor and suggest tumor progression from benign to borderline and from borderline to carcinoma. Therefore, extensive sampling for histological examination is necessary [49]. The category of mucinous borderline tumor with intraepithelial carcinoma is used for those tumors that lack obvious stromal invasion but show areas, less than 10 mm2, where the cytological features of the tumor cells are unequivocally malignant [50]. Mucinous borderline tumors with intraepithelial carcinoma have a very low risk of recurrence, of less than 5% [51].
Recently, MCs have been divided into two categories: (a) an expansile type without obvious stromal invasion, but exhibiting back-to-back or complex malignant glands with minimal or no intervening stroma, and exceeding 10 mm2 in area (>3 mm in each of two linear dimensions) (Fig. 1c); and (b) an infiltrative type, showing evident stromal invasion in the form of glands, cell clusters, or individual cells, disorderly infiltrating the stroma and frequently associated with a desmoplastic stromal reaction [49, 50]. The expansile pattern of growth has also been referred to as the “noninvasive”, “intraglandular”, [52] or “confluent glandular” [53] pattern and is associated with a more favorable prognosis than the infiltrative pattern. A histopathological feature unique to mucinous tumors is the occasional finding of mural nodules of anaplastic carcinoma or high-grade sarcoma. When such nodules are localized in the wall of an unruptured cyst, the prognosis may be favorable, but such tumors may recur and do so as the anaplastic component [49, 54].
The gene expression profile of MCs differs from those of serous, endometrioid, and clear cell carcinomas [55]. As expected, MCs have genes that encode markers of intestinal differentiation such as CDX2 and KRAS. KRAS mutations, which are an early event in mucinous tumorigenesis, are frequent in ovarian MCs [56]. Primary ovarian mucinous tumors are almost always (up to 80%) immunoreactive for cytokeratin 7 (CK7) whereas colorectal adenocarcinomas are usually CK7 negative [57]. Ovarian MCs are immunoreactive for CK20 in 65% of cases, but the reaction is typically weak and focal; staining for CDX-2 (nuclear immunoreaction) is similar [58]. In contrast, colorectal adenocarcinomas are diffusely and strongly reactive for CK20 and CDX-2. Loss of Dpc4 immunoreactivity occurs in almost 50% of metastatic carcinomas of the pancreas, whereas most primary ovarian MCs are focally or diffusely positive [59]. Human papilloma virus (HPV) DNA assessment may be helpful for distinguishing mucinous adenocarcinoma of the cervix metastatic to the ovary from a primary ovarian MC. p16 expression is also a reliable surrogate marker for HPV [60]. MCs are uniformly negative for ER and WT1, in contrast to endometrioid (ER+) and serous (ER + and WT1+) carcinomas.
Endometrioid carcinomas
Endometrioid tumors of the ovary closely mimic their uterine counterparts. ECs account for 10% of all ovarian carcinomas, occur most frequently in women of perimenopausal age, and most are found at an early stage [1]. The ovarian tumors are bilateral in 28% of cases and are associated in 15–20% of cases with carcinoma of the endometrium [2, 61]. Most ECs are low-grade adenocarcinomas and seem to arise from endometriotic cysts. Up to 42% of cases have evidence of ipsilateral ovarian or pelvic endometriosis [1, 2]. Squamous differentiation occurs in 50% of cases [1] (Fig.1d). In contrast, high-grade ECs are morphologically indistinguishable from HGSCs and often express WT1. Gene expression profiling is also similar, suggesting that high-grade EC is not a distinct tumor type [62].
It has been recognized that atypical endometriosis is the precursor lesion of endometrioid and clear cell carcinomas of the ovary, and a direct transition from ovarian atypical endometriosis to endometrioid or clear cell carcinomas has been described in 15–32% of cases [63] (Fig. 5). In cases of ovarian endometrioid carcinoma associated with endometriosis, common genetic alterations have been encountered in the adjacent endometriosis, atypical endometriosis, and adenocarcinoma [64]. In mice harboring KRAS mutations that result in the development of benign lesions reminiscent of endometriosis, deletion of PTEN leads to the induction of invasive endometrioid carcinoma [65]. These results indicate that inactivation of tumor suppressor genes such as PTEN may represent early events in the malignant transformation of endometriosis [20].

Endometrioid carcinoma arising from endometriosis. The inset shows squamous differentiation
With the recent discovery of AT-rich interactive domain 1A gene (ARID1A) mutations in endometrioid and clear cell carcinomas as well as in adjacent endometriosis, there is renewed interest in the molecular events that occur in precursor lesions [66]. ARID1A is a component of the SWI–SNF-A complex, a large, multiprotein chromatin remodeling complex which is known to both enhance and repress transcription [67]. It behaves as a tumor suppressor gene. BAF250 protein, encoded by ARID1A, plays a crucial role in chromatin remodeling. The question has been raised whether endometriotic lesions should be analyzed for expression of BAF250. Also, if patients with endometriotic lesions that show loss of expression should be viewed as being at high risk for the development of CCC or endometrioid ovarian cancers [68].
Somatic mutations of the beta-catenin (CTNNB1) and PTEN genes are the most common genetic abnormalities identified in ovarian ECs [19, 69, 70]. Compared with uterine ECs, ovarian ECs have a similar frequency of beta-catenin abnormalities but lower rate of microsatellite instability (MI) and PTEN alterations [70]. CTNNB1 mutations, which occur in 38%–50% of cases, are associated with squamous differentiation, low tumor grade, and favorable outcome [71]. Mutations have been described in exon 3 (codons 32, 33, 37, and 41) and involve the phosphorylation sequence for glycogen synthase kinase 3-beta. These mutations probably render a fraction of cellular beta-catenin insensitive to APC-mediated downregulation and are responsible for its accumulation in the nuclei of the tumor cells. Beta-catenin is immunohistochemically detectable in carcinoma cells in more than 80% of the cases.
PTEN inactivation results in activation of the PI3K-AKT signaling pathway that inhibits apoptosis. PTEN is mutated in approximately 20% of ovarian ECs and in 46% of those with 10q23 LOH. PTEN mutations occur between exons 3 and 8. The finding of LOH at 10q23 and somatic PTEN mutations in endometriotic cysts adjacent to ECs with similar genetic alterations provides additional evidence for the precursor role of endometriosis in ovarian carcinogenesis [20]. An alternative mechanism for activation of the PI3K signaling in ECs is through activating mutations of PIK3CA, which encodes the p110 catalytic subunit of PI3K. PIK3CA mutations in exons 9 and 20 have been identified in 20% of ovarian ECs and CCCs but in only 2% of HGSC [72]. PIK3CA mutations are associated with adverse prognostic parameters [73, 74].
ECs are the types most commonly encountered in patients with hereditary non-polyposis colon cancer syndrome. The reported frequency of MI in ovarian ECs ranges from 12.5% to 19% [75, 76]. Like endometrial carcinomas, ovarian ECs with MI follow the same process of MLH-1 promoter methylation and frameshift mutations at coding mononucleotide repeat microsatellites [70]. ECs are immunoreactive for vimentin, cytokeratins (CK7, 97%; CK20, 13%), epithelial membrane antigen, and estrogen and progesterone receptors. Immunoreaction for alpha-inhibin, WT-1, and calretinin are negative in most ECs. The median Ki-67 labeling index for endometrioid is 8.2% [10].
Simultaneous carcinomas of the uterine corpus and ovary occur in 15–20% of ovarian tumors and in approximately 5% of uterine tumors [2]. Both tumors are of endometrioid type in the majority of cases. In addition to prognostic implications, accurate diagnosis as independent primaries or metastases is necessary for appropriate staging and treatment. Assessment of conventional pathological features including tumor size, histological type and grade, pattern of tumor growth, vascular invasion, and coexisting atypical hyperplasia or endometriosis allows the distinction between primary and metastatic tumors in most cases [2]. Occasionally, however, the differential diagnosis can be difficult or impossible as the tumors may show overlapping features [2]. In such cases, patient follow-up is the single most conclusive factor, but ancillary techniques may help to establish the correct diagnosis.
Clonality analysis using LOH and gene mutation is useful in the distinction of independent primary carcinomas from metastatic carcinomas, provided the diagnosis does not rely exclusively on a single molecular result and the molecular data are interpreted in the light of appropriate clinical and pathologic findings [61]. According to a recent study [61], the frequency of molecular alterations in both independent and metastatic tumors, including MI and PTEN mutations, is higher than that observed in single sporadic tumors. Nuclear immunoreactivities for β-catenin and CTNNB1 mutations were restricted to independent uterine and ovarian tumors and were absent in metastatic tumors. These findings correlated with the clinical outcome [61].
EC is the type of ovarian carcinoma with the most favorable prognosis. Although their lower grade and lower stage account for much of the favorable prognosis, responsiveness to chemotherapy may also be a contributory factor.
Clear cell carcinomas
CCCs account for approximately 10% of ovarian carcinomas and patients typically present with stage 1 or 2 disease. Tumors are rarely bilateral. CCCs are associated with an unfavorable prognosis when they present at advanced stage [77]. As with EC, there is an association with endometriosis, and CCCs associated with endometriosis have a favorable prognosis [78].
The presence of clear cells alone is not sufficient for a diagnosis of CCC, as cells with clear cytoplasm can be seen in HGSC and EC. Besides the characteristic clear or hobnail cells with eccentric, rounded, and bulbous nuclei, the diagnosis is based on the following architectural and cytological findings: (a) multiple complex papillae; (b) densely hyaline basement membrane material expanding the cores of the papillae (Fig. 1e); and (c) hyaline bodies, which are present in approximately 25% of cases. Mitoses are less frequent than in other types of ovarian carcinomas (usually less than 5 /10 HPFs).
CCCs lack the BRCA abnormalities, chromosomal instability, or complex karyotypes of HGSC [79]. Recently, it has been found that nearly half the CCCs (46–57%) carry ARID1A mutations and lack BAF250 protein [66]. In two cases, ARID1A mutations and loss of BAF250a expression were found in the tumor and adjacent endometriosis but not in distant endometriosis. This finding suggests that ARID1A inactivation occurs early during malignant transformation of endometriosis [66]. CCCs are usually positive for HNF1-beta (>90%) and are negative for ER and WT1 in more than 95% of cases [10, 28].
Hepatocyte nuclear factor-1beta (HNF-1beta) is upregulated in ovarian clear cell tumors, including benign, borderline tumors, and carcinomas [80]. This transcription factor facilitates glycogen synthesis and is expressed in mid-to-late secretory and gestational endometrium (Arias–Stella reaction) [80], atypical and inflammatory endometriosis, and clear cell carcinoma [80]. HNF-1beta regulates several specific genes of clear cell carcinoma, including dipeptidyl peptidase IV (glycogen synthesis), osteopontin (progesterone-regulated endometrial secretory protein), angiotensin converting enzyme 2 (ferritin induction, iron deposition, antiapoptosis), annexin 4 (paclitaxel resistance), and UGT1A1 (detoxification) [81]. Thus, HNF-1beta appears to play an important role in the pathogenesis and behavior of clear cell carcinoma.
The RHO GTPase family of proteins is involved in tumor progression through cytoskeleton regulation [82]. Compared with HGSCs, CDC42 mRNA levels are significantly lower in CCCs. In contrast, the expression of Rho GDP dissociation inhibitor gamma (ARHGDIG) mRNA is higher in CCCs than HGSCs. In patients with clear cell carcinoma, high expression of ARHGDIG mRNA is associated with low stage, fewer recurrences, and better survival [83]. Thus, RHO GTPase inhibition could explain the fact that clear cell carcinomas of the ovary are found at stage I in about 25% of cases.
CCCs are less likely to respond to chemotherapy than HGSCs [8]. The reported differences in response rates (15–45%) may reflect inclusion of HGSC with clear cell change in some case series. Whereas highly proliferative cells that lack the ability to repair double-stranded DNA (i.e., HGSC cells) show sensitivity to platinum-based chemotherapy, the less proliferative, genomically stable cells of CCC are less sensitive to platinum compounds [84].
Mixed epithelial tumors
As noted previously, if tumors with an undifferentiated component are classified based on the areas showing a recognizable growth pattern, they usually will end up classified as HGSC. Most tumors considered in the past to be mixed high-grade serous/endometrioid, high-grade serous/clear cell, or high-grade serous/transitional cell are, based on molecular studies, better classified as HGSC. One of the most common mixed epithelial carcinomas of ovary are EC/CCC. These tumors are often associated with endometriosis and, based on the presence of a CCC component, it is appropriate to consider them high-grade carcinomas.
Conclusions
The five main types of ovarian carcinoma (in descending order of frequency: HGSC, CCC, EC, MC, and LGSC) account for 98% of ovarian carcinomas, can be reproducibly diagnosed, and are inherently different diseases, as indicated by differences in risk factors, molecular genetic abnormalities, natural history, and response to chemotherapy. Even if HGSC comprise 70% of the total, lumping the other four types together for classifying ovarian carcinomas into just two types (“types I and II”) is artificial and limits progress in understanding the biology or improving the treatment of the less common types of ovarian carcinomas. For a successful type-specific treatment of ovarian carcinoma, reproducible histopathological diagnosis of the tumor cell type is critical.
Even if a mesothelial origin cannot be excluded, there is compelling evidence that a significant number of cases of HGSC originate in the distal fallopian tube. The identification of a precursor lesion (TIC) within the fallopian tube fimbria opens new ways for prevention in the general population. Given that a significant number of “ovarian cancers” originate in the fallopian tube, removal during surgery of the fallopian tubes while keeping the hormone producing ovaries in place would prevent the adverse effects of early menopause such as osteoporosis or vasomotor symptoms.
Although endometriosis is essentially a benign disease, it does exhibit some features reminiscent of malignancy such as metastatic potential and monoclonality. Whereas the incidence of malignant tumors arising from ovarian endometriosis is minimal, the incidence of endometriosis in women with ovarian cancer is more significant. It has been recognized that atypical endometriosis is the precursor lesion of EC and CCC of the ovary and a direct transition from ovarian atypical endometriosis to EC or CCC has been described. The question has been raised whether the finding in endometriotic lesions of the genetic alterations frequently encountered in EC and CCC should be interpreted as evidence of high risk of malignant transformation.
References
Prat J (2004) Pathology of the ovary. Saunders, Philadelphia
Lee KR, Tavassoli FA, Prat J et al (2003) Surface epithelial-stromal tumours (Ch 2: tumours of the ovary and peritoneum). In: Tavassoli FA, Devilee P (eds) World Health Organization classification of tumours: pathology and genetics of tumours of the breast and female genital organs. IARC Press, Lyon, pp 117–145
Shvartsman HS, Sun CC, Bodurka CC, Mahajan V, Crispens M, Lu KH, Deavers MT, Malpica A, Silva EG, Gershenson DM (2007) Comparison of the clinical behavior of newly diagnosed stages II–IV low-grade serous carcinoma of the ovary with that of serous ovarian tumors of low malignant potential that recur as low-grade serous carcinoma. Gynecol Oncol 105:625–629
Stewart BW, Kleihues P (2003). World Cancer Report (IARC Press)
Jass JR (2006) Colorectal cancer: a multipathway disease. Crit Rev Oncog 12:273–287
Gilks CB, Prat J (2009) Ovarian carcinoma pathology and genetics: recent advances. Hum Pathol 40:1213–1223
Takano M, Kikuchi Y, Yaegashi N, Kuzuya K, Ueki M, Tsuda H, Suzuki M, Kigawa J, Takeuchi S, Tsuda H, Moriya T, Sugiyama T (2006) Clear cell carcinoma of the ovary: a retrospective multicentre experience of 254 patients with complete surgical staging. Br J Cancer 94:1369–1374
du Bois A, Lück HJ, Meier W, Adams HP, Möbus V, Costa S, Bauknecht T, Richter B, Warm M, Schröder W, Olbricht S, Nitz U, Jackisch C, Emons G, Wagner U, Kuhn W, Pfisterer J (2003) A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J Natl Cancer Inst 95:1320–1329
Fountain J, Trimble E, Birrer MJ (2006) Summary and discussion of session recommendations. Gynecol Oncol 103:S23–S25
Köbel M, Kalloger SE, Boyd N, McKinney S, Mehl E, Palmer C, Leung S, Bowen NJ, Ionescu DN, Rajput A, Prentice LM, Miller D, Santos J, Swenerton K, Gilks CB, Huntsman D (2008) Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med 5:e232
Kurman RJ, Shih IM (2010) The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol 34:433–443
Piek JM, van Diest PJ, Zweemer RP, Jansen JW, Poort-Keesom RJ, Menko FH, Gille JJ, Jongsma AP, Pals G, Kenemans P, Verheijen RH (2001) Dysplastic changes in prophylactically removed fallopian tubes of women predisposed to developing ovarian cancer. J Pathol 195:451–456
Piek JM, van Diest PJ, Zweemer RP, Kenemans P, Verheijen RH (2001) Tubal ligation and risk of ovarian cancer. Lancet 358:844
Medeiros F, Muto MG, Lee Y, Elvin JA, Callahan MJ, Feltmate C, Garber JE, Cramer DW, Crum CP (2006) The tubal fimbria is a preferred site for early adenocarcinoma in women with familial ovarian cancer syndrome. Am J Surg Pathol 30:230–236
Kindelberger DW, Lee Y, Miron A, Hirsch MS, Feltmate C, Medeiros F, Callahan MJ, Garner EO, Gordon RW, Birch C, Berkowitz RS, Muto MG, Crum CP (2007) Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: evidence for a causal relationship. Am J Surg Pathol 31:161–169
Lee Y, Miron A, Drapkin R, Nucci MR, Medeiros F, Saleemuddin A, Garber J, Birch C, Mou H, Gordon RW, Cramer DW, McKeon FD, Crum CP (2007) A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J Pathol 211:26–35
Folkins AK, Jarboe EA, Saleemuddin A, Lee Y, Callahan MJ, Drapkin R, Garber JE, Muto MG, Tworoger S, Crum CP (2008) A candidate precursor to pelvic serous cancer (p53 signature) and its prevalence in ovaries and fallopian tubes from women with BRCA mutations. Gynecol Oncol 109:168–173
Jarboe E, Folkins A, Nucci MR, Kindelberger D, Drapkin R, Miron A, Lee Y, Crum CP (2008) Serous carcinogenesis in the fallopian tube: a descriptive classification. Int J Gynecol Pathol 27:1–9
Obata K, Morland SJ, Watson RH, Hitchcock A, Chenevix-Trench G, Thomas EJ, Campbell IG (1998) Frequent PTEN/MMAC mutations in endometrioid but not serous or mucinous epithelial ovarian tumors. Cancer Res 58:2095–2097
Sato N, Tsunoda H, Nishida M, Morishita Y, Takimoto Y, Kubo T, Noguchi M (2000) Loss of heterozygosity on 10q23.3 and mutation of the tumor suppressor gene PTEN in benign endometrial cyst of the ovary: possible sequence progression from benign endometrial cyst to endometrioid carcinoma and clear cell carcinoma of the ovary. Cancer Res 60:7052–7056
Singer G, Oldt R 3rd, Cohen Y, Wang BG, Sidransky D, Kurman RJ, IeM S (2003) Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Nat Cancer Inst 95:484–486
Singer G, Stöhr R, Cope L, Dehari R, Hartmann A, Cao DF, Wang TL, Kurman RJ, IeM S (2005) Patterns of p53 mutations separate ovarian serous borderline tumors and low- and high-grade carcinomas and provide support for a new model of ovarian carcinogenesis. Am J Surg Pathol 29:218–224
Laury AR, Ning G, Quick CM, Nucci MR, Parast MM, McKeon FD, Xian W, Crum CP (2011) Fallopian tube correlates of serous borderline tumors. Am J Surg Pathol 35:1759–1765
Kurman RJ, Vang R, Junge J, Hannibal CG, Kjaer SK, Shih Ie M (2011) Papillary tubal hyperplasia: the putative precursor of ovarian atypical proliferative (borderline) serous tumors, noninvasive implants, and endosalpingiosis. Am J Surg Pathol 35:1605–1614
Malpica A, Deavers MT, Lu K, Bodurka DC, Atkinson EN, Gershenson DM, Silva EG (2004) Grading ovarian serous carcinoma using a two-tier system. Am J Surg Pathol 28:496–504
Gilks CB, Ionescu DN, Kalloger SE, Köbel M, Irving J, Clarke B, Santos J, Le N, Moravan V, Swenerton K (2008) Tumor cell type can be reproducibly diagnosed and is of independent prognostic significance in patients with maximally debulked ovarian carcinoma. Hum Pathol 39:1239–1251
Al-Hussaini M, Stockman A, Foster H, Mc Cluggage WG (2004) WT-1 assists in distinguishing ovarian from uterine serous carcinoma and in distinguishing between serous and endometrioid ovarian carcinoma. Histopathology 44:109–105
Köbel M, Kalloger SE, Carrick J, Huntsman D, Asad H, Oliva E, Ewanowich CA, Soslow RA, Gilks CB (2009) A limited panel of immunomarkers can reliably distinguish between clear cell and high-grade serous carcinoma of the ovary. Am J Surg Pathol 33:14–21
Karst AM, Levanon K, Duraisamy S, Liu JF, Hirsch MS, Hecht JL, Drapkin R (2011) Stathmin 1, a marker of PI3K pathway activation and regulator of microtubule dynamics, is expressed in early pelvic serous carcinomas. Gynecol Oncol 123:5–12
Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Fan I, Tang J, Li S, Zhang S, Shaw PA, Narod SA (2006) Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: a kin-cohort study in Ontario, Canada. J Natl Cancer Inst 98:1694–1706
Venkitaraman AR (2009) Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu Rev Pathol 4:461–487
Geisler JP, Hatterman-Zogg MA, Rathe JA, Buller RE (2002) Frequency of BRCA1 dysfunction in ovarian cancer. J Natl Cancer Inst 94:61–67
Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Prat J, Harkes I, Repasky EA, Gabrielson E, Schutte M, Baylin SB, Herman JG (2000) Promoter hypermethylation is a cause of BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst 92:564–569
Bowtell DD (2010) The genesis and evolution of high-grade serous ovarian cancer. Nat Rev Cancer 10:803–808
Levanon K, Ng V, Piao HY, Zhang Y, Chang MC, Roh MH, Kindelberger DW, Hirsch MS, Crum CP, Marto JA, Drapkin R (2010) Primary ex vivo cultures of human fallopian tube epithelium as a model for serous ovarian carcinogenesis. Oncogene 29:1103–1113
Powell CB, Kenley E, Chen LM, Crawford B, McLennan J, Zaloudek C, Komaromy M, Beattie M, Ziegler J (2005) Risk-reducing salpingo-oophorectomy in BRCA mutation carriers: role of serial sectioning in the detection of occult malignancy. J Clin Oncol 23:127–132
Pothuri B, Leitao MM, Levine DA, Viale A, Olshen AB, Arroyo C, Bogomolniy F, Olvera N, Lin O, Soslow RA, Robson ME, Offit K, Barakat RR, Boyd J (2010) Genetic analysis of the early natural history of epithelial ovarian carcinoma. PLoS One 5:e10358
Norquist BM, Garcia RL, Allison KH, Jokinen CH, Kernochan LE, Pizzi CC, Barrow BJ, Goff BA, Swisher EM (2010) The molecular pathogenesis of hereditary ovarian carcinoma: alterations in the tubal epithelium of women with BRCA1 and BRCA2 mutations. Cancer 116:5261–5271
Espinosa I, Catasus L, Canet B, D’Angelo E, Muñoz J, Prat J (2011) Gene expression analysis identifies two groups of ovarian high-grade serous carcinomas with different prognosis. Mod Pathol 24:846–854
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A (2005) Targeting the DNA repair defectin BRCA mutant cells as a therapeutic strategy. Nature 434:917–921
Tutt AN, Lord CJ, McCabe N, Farmer H, Turner N, Martin NM, Jackson SP, Smith GC, Ashworth A (2005) Exploiting the DNA repair defectin BRCA mutant cells in the design of new therapeutic strategies for cancer. Cold Spring Harb Symp Quant Biol 70:139–148
Gershenson DM, Sun CC, Lu KH, Coleman RL, Sood AK, Malpica A, Deavers MT, Silva EG, Bodurka DC (2006) Clinical behavior of stage II–IV low-grade serous carcinoma of the ovary. Obstet Gynecol 108:361–368
Malpica A, Deavers MT, Tornos C, Kurman RJ, Soslow R, Seidman JD, Munsell MF, Gaertner E, Frishberg D, Silva EG (2007) Inter-observer and intraobserver variability of a two-tier system for grading ovarian serous carcinoma. Am J Surg Pathol 31:1203–1208
Ayhan A, Kurman RJ, Yemelyanova A, Vang R, Logani S, Seidman JD, IeM S (2009) Defining the cut-point between low- and high-grade ovarian serous carcinomas: a clinicopathologic and molecular genetic analysis. Am J Surg Pathol 33:1220–1224
Jones S, Wang T-L, Kurman RJ, Nakayama K, Velculescu VE, Vogelstein B, Kinzler KW, Papadopoulos N, Shih I-M (2012) Low-grade serous carcinomas of the ovary contain very few point mutations. J Pathol 226:413–420
Bell DA, Longacre TA, Prat J, Kohn EC, Soslow RA, Ellenson LH, Malpica A, Stoler MH, Kurman RJ (2004) Serous borderline (low malignant potential, atypical proliferative) ovarian tumors: workshop perspectives. Hum Pathol 35:934–948
Sieben NL, Roemen GMJM, Oosting J, Fleuren GJ, van Engeland M, Prat J (2006) Clonal analysis favours a monoclonal origin for serous borderline tumours with peritoneal implants. J Pathol 210:405–411
Crispens MA, Bodurka D, Deavers M, Lu K, Silva EG, Gershenson DM (2002) Response and survival in patients with progressive or recurrent serous ovarian tumors of low malignant potential. Obstet Gynecol 99:3–10
Rodriguez IM, Prat J (2002) Mucinous tumors of the ovary: a clinicopathologic analysis of 75 borderline tumors (of intestinal type) and carcinomas. Am J Surg Pathol 26:139–152
Lee KR, Scully RE (2000) Mucinous tumors of the ovary—A clinicopathologic study of 196 borderline tumors (of intestinal type) and carcinomas, including an evaluation of 11 cases with “pseudomyxomaperitonei”. Am J Surg Pathol 24:1447–1464
Silverberg SG, Bell DA, Kurman RJ, Seidman JD, Prat J, Ronnett BM, Copeland L, Silva E, Gorstein F, Young RH (2004) Borderline ovarian tumors: key points and workshop summary. Hum Pathol 35:910–917
Hoerl HD, Hart WR (1998) Primary ovarian mucinous cystadenocarcinomas: a clinicopathologic study of 49 cases with long term follow-up. Am J Surg Pathol 22:1449–1462
Riopel MA, Ronnett BM, Kurman RJ (1999) Evaluation of diagnostic criteria and behavior of ovarian intestinal-type mucinous tumors. Atypical proliferative (borderline) tumors and intraepithelial, microinvasive, invasive and metastatic carcinomas. Am J Surg Pathol 23:617–635
Provenza C, Young RH, Prat J (2008) Anaplastic carcinoma in mucinous ovarian tumors: a clinicopathologic study of 34 cases emphasizing the crucial impact of stage on prognosis, their histologic spectrum, and overlap with sarcoma-like mural nodules. Am J Surg Pathol 32:383–389
Heinzelmann-Schwarz VA, Gardiner-Garden M, Henshall SM, Scurry JP, Scolyer RA, Smith AN, Bali A, Vanden Bergh P, Baron-Hay S, Scott C, Fink D, Hacker NF, Sutherland RL, O’Brien PM (2006) A distinct molecular profile associated with mucinous epithelial ovarian cancer. Br J Cancer 94:904–91353
Cuatrecasas M, Villanueva A, Matias-Guiu X, Prat J (1997) K-ras mutations in mucinous ovarian tumors. A clinicopathologic and molecular study of 95 cases. Cancer 79:1581–1586
Park SY, Kim HS, Hong EK, Kim WH (2002) Expression of cytokeratins 7 and 20 in primary carcinomas of the stomach and colorectum and their value in the differential diagnosis of metastatic carcinomas to the ovary. Hum Pathol 33:1078–1085
Vang R, Gown AM, Wu LS, Barry TS, Wheeler DT, Yemelyanova A, Seidman JD, Ronnett BM (2006) Immunohistochemical expression ofCDX2 in primary ovarian mucinous tumors and metastatic mucinous carcinomas involving the ovary: comparison with CK20 and correlation with coordinate expression of CK7. Mod Pathol 19:1421–1428
Ji H, Isacson C, Seidman J, Kurman RJ, Ronnett BM (2002) Cytokeratins 7and 20, Dpc4, and MUC5AC in the distinction of metastatic mucinous carcinomas in the ovary from primary ovarian mucinous tumors: Dpc4assists in identifying metastatic pancreatic carcinomas. Int J Gynecol Pathol 21:391–400
Ronnett BM, Yemelyanova AV, Vang R, Gilks CB, Miller D, Gravitt PE, Kurman RJ (2008) Endocervical adenocarcinomas with ovarian metastases: analysis of 29 cases with emphasis on minimally invasive cervical tumors and the ability of the metastases to simulate primary ovarian neoplasms. Am J Surg Pathol 32:1835–1853
Irving JA, Catasús L, Gallardo A, Bussaglia E, Romero M, Matias-Guiu X, Prat J (2005) Synchronous endometrioid carcinomas of the uterine corpus and ovary: alterations in the beta-catenin (CTNNB1) pathway are associated with independent primary tumors and favorable prognosis. Hum Pathol 36:605–619
Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, Johnson DS, Trivett MK, Etemadmoghadam D, Locandro B, Traficante N, Fereday S, Hung JA, Chiew YE, Haviv I, Australian Ovarian Cancer Study Group, Gertig D, DeFazio A, Bowtell D (2008) Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res 14:5198–5208
Sainz de la Cuesta R, Eichhorn JH, Rice LW, Fuller AF Jr, Nikrui N, Goff BA (1996) Histologic transformation of benign endometriosis to early epithelial ovarian cancer. Gynecol Oncol 60:238–244
Jiang X, Morland SJ, Hitchcock A, Thomas EJ, Campbell IG (1998) Allelotyping of endometriosis with adjacent ovarian carcinoma reveals evidence of a common lineage. Cancer Res 58:1707–1712
Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T (2005) Role of K-ras and PTEN in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med 11:63–70
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, Yang W, Heravi-Moussavi A, Giuliany R, Chow C, Fee J, Zayed A, Prentice L, Melnyk N, Turashvili G, Delaney AD, Madore J, Yip S, McPherson AW, Ha G, Bell L, Fereday S, Tam A, Galletta L, Tonin PN, Provencher D, Miller D, Jones SJ, Moore RA, Morin GB, Oloumi A, Boyd N, Aparicio SA, Shih Ie M, Mes-Masson AM, Bowtell DD, Hirst M, Gilks B, Marra MA, Huntsman DG (2010) ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 363:1532–1543
Lemon B, Inouye C, King DS, Tjian R (2001) Selectivity of chromatin-remodelling cofactors for ligand-activated transcription. Nature 414:924–928
Birrer MJ (2010) The origin of ovarian cancer—is it getting clearer? N Engl J Med 363:1574–1575
Palacios J, Gamallo C (1998) Mutations in the beta-catenin gene (CTNNB1) in endometrioid ovarian carcinomas. Cancer Res 58:1344–1347
Catasús L, Bussaglia E, Rodriguez I, Gallardo A, Pons C, Irving JA, Prat J (2004) Molecular genetic alterations in endometrioid carcinomas of the ovary: similar frequency of beta-catenin abnormalities but lower rate of microsatellite instability and PTEN alterations than in uterine endometrioid carcinomas. Hum Pathol 35:1360–1368
Gamallo C, Palacios J, Moreno G, Calvo de Mora J, Suárez A, Armas A (1999) Beta-catenin expression pattern in stage I and II ovarian carcinomas: relationship withbeta-catenin gene mutations, clinicopathological features, and clinical outcome. Am J Pathol 155:527–536
Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, Cristiano BE, Pearson RB, Phillips WA (2004) Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res 64:7678–7681
Catasus L, Gallardo A, Cuatrecasas M, Prat J (2008) PIK3CA mutations in the kinase domain (exon 20) of uterine endometrial adenocarcinomas are associated with adverse prognostic parameters. Mod Pathol 21:131–139
Willner J, Wurz K, Allison KH, Galic V, Garcia RL, Goff BA, Swisher EM (2007) Alternate molecular genetic pathways in ovarian carcinomas of common histological types. Hum Pathol 38:607–613
Gras E, Catasus L, Argüelles R, Moreno-Bueno G, Palacios J, Gamallo C, Matias-Guiu X, Prat J (2001) Microsatellite instability, MLH-1 promoter hypermethylation, and frameshift mutations at coding mononucleotide repeat microsatellites in ovarian tumors. Cancer 92:2829–2836
Moreno-Bueno G, Gamallo C, Perez-Gallego L, Calvo de Mora J, Suarez A, Palacios J (2001) Beta-catenin expression pattern, beta-catenin gene mutations, and microsatellite instability in endometrioid ovarian carcinomas and synchronous endometrial carcinomas. Diagn Mol Pathol 10:116–122
Sugiyama T, Kamura T, Kigawa J, Terakawa N, Kikuchi Y, Kita T, Suzuki M, Sato I, Taguchi K (2000) Clinical characteristics of clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum based chemotherapy. Cancer 88:2584–2589
Komiyama S, Aoki D, Tominaga E, Susumu N, Udagawa Y, Nozawa S (1999) Prognosis of Japanese patients with ovarian clear cell carcinoma associated with pelvic endometriosis: clinicopathologic evaluation. Gynecol Oncol 72:342–346
Press JZ, De Luca A, Boyd N, Young S, Troussard A, Ridge Y, Kaurah P, Kalloger SE, Blood KA, Smith M, Spellman PT, Wang Y, Miller DM, Horsman D, Faham M, Gilks CB, Gray J, Huntsman DG (2008) Ovarian carcinomas with genetic and epigenetic BRCA1 loss have distinct molecular abnormalities. BMC Cancer 8:17
Kato N, Sasou S, Motoyama T (2006) Expression of hepatocyte nuclear factor-1beta (HNF-1beta) in clear cell tumors and endometriosis of the ovary. Mod Pathol 19:83–89
Kobayashi H, Yamada Y, Kanayama S, Furukawa N, Noguchi T, Haruta S, Yoshida S, Sakata M, Sado T, Oi H (2009) The role of hepatocyte nuclear factor-1beta in the pathogenesis of clear cell carcinoma of the ovary. Int J Gynecol Cancer 19:471–479
Horiuchi A, Imai T, Wang C, Ohira S, Feng Y, Nikaido T, Konishi I (2003) Upregulation of small GTPases, RhoA and RhoC, is associated with tumor progression in ovarian carcinoma. Lab Invest 83:861–870
Canet B, Pons C, Espinosa I, Prat J (2011) Ovarian clear cell carcinomas: RHO GTPases may contribute to explain their singular biologic behavior. Hum Pathol 42:833–839
Itamochi H, Kigawa J, Sugiyama T, Kikuchi Y, Suzuki M, Terakawa N (2002) Low proliferation activity may be associated with chemoresistance in clear cell carcinoma of the ovary. Obstet Gynecol 100:281–287
Conflicts of interest
The author declares that there are no conflicts of interest
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Prat, J. Ovarian carcinomas: five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch 460, 237–249 (2012). https://doi.org/10.1007/s00428-012-1203-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-012-1203-5