

Abstract
Background/Aim: Invariant natural killer T (iNKT) cells are involved in the initiation and resolution of inflammatory responses. We previously reported that activated iNKT cells facilitate liver repair after hepatic ischemia reperfusion (I/R) injury by accelerating macrophage polarization during the early phase of hepatic I/R injury. Upon activation with α-galactosylceramide (α-GalCer), iNKT cell numbers transiently decrease before increasing within 72 h of stimulation. In the present study, we examined the role of expanded hepatic iNKT cells in the late phase of hepatic I/R injury. Materials and Methods: iNKT cells were activated by intraperitoneal injection of α-GalCer in male C57/BL6 mice at the induction of hepatic ischemia followed by reperfusion. Results: Numbers of activated hepatic iNKT cells immediately diminished after hepatic I/R and reached minimal levels at 24 h and 48 h post-reperfusion. Numbers of hepatic iNKT cells then increased at 72 h and 96 h post-reperfusion to levels approximately 2-fold higher than in mice that underwent a sham operation. Liver repair as demonstrated by decreased necrotic area and increased expression of proliferating cell nuclear antigen (PCNA) was enhanced in α-GalCer-treated mice at 96 h post-reperfusion. Interleukin (IL)-13 production by proliferating iNKT cells was observed at 96 h post-reperfusion, which was associated with enhanced liver repair and increased numbers of reparative macrophages. Conclusion: Repopulation of hepatic iNKT cells promotes liver repair by stimulating macrophage phenotype switching in the late phase of hepatic I/R injury.
- Liver
- recovery
- iNKT cells
- pro-inflammatory macrophages
- reparative macrophages
Despite recent advances in liver surgical devices and techniques, hepatic ischemia reperfusion (I/R) injury remains an inevitable complication of liver resection and liver transplantation. Delays in the process of liver repair and regeneration after hepatic I/R injury result in a significantly increased risk of postoperative morbidity and mortality (1). Macrophages are a key player in recovery from hepatic I/R injury (2-4). Macrophages can be functionally classified into pro-inflammatory macrophages and reparative macrophages according to responses to different stimuli. Macrophage polarization refers to phenotype switching of macrophages from pro-inflammatory to reparative phenotypes at the site of injury. Accumulation of macrophages at the sites of injury followed by macrophage polarization contributes to liver repair after acute liver injury (5). However, the mechanisms underlying the induction of macrophage phenotypic switching have yet to be fully elucidated.
Invariant natural killer T (iNKT) cells recognize glycolipid antigens expressed on the surface molecule, CD1d, by the invariant T cell receptor (TCR). The reactivity of iNKT cells is restricted by CD1d (6). Upon activation, iNKT cells secrete large amounts of cytokines, including interferon (IFN)-γ and interleukin (IL)-4, as well as chemokines which modulate subsequent immune responses (7, 8). A specific ligand for iNKT cells, α-galactosylceramide (α-GalCer) (9), binds to CD1d leading to extensive activation of iNKT cells (10). In liver, activated iNKT cells can have protective or deleterious roles during hepatic I/R injury. iNKT cells are involved in the initiation of hepatic I/R injury (11, 12), while pre-activation of iNKT cells by α-GalCer attenuates inflammation induced by hepatic I/R (13). In addition, recent studies have posited that iNKT cells contribute to the resolution of inflammation and tissue repair after I/R in the heart (14) and after focal thermal trauma in the liver (15). Furthermore, we have recently reported that liver repair after hepatic I/R injury was promoted by the accelerated polarization of macrophages through interaction with activated iNKT cells stimulated with α-GalCer (16). Activated iNKT cells were shown to produce both IL-4 and IFN-γ in the early phase of hepatic I/R injury (6 h post-reperfusion). IFN-γ induced rapid accumulation of pro-inflammatory macrophages and IL-4 induced macrophage phenotypic switching from pro-inflammatory to reparative phenotypes contributing to repair of tissue injury induced by hepatic I/R (16). These findings indicate that iNKT cells interact with macrophages to facilitate the resolution of hepatic inflammation and repair after acute liver injury (17); however, few studies have examined the role of crosstalk between iNKT cells and macrophages during tissue repair after injury (18).
Of interest, the administration of α-GalCer alone resulted in a rapid reduction in the number of hepatic iNKT cells within 8 h of administration through down-regulation of iNKT TCR (19). iNKT cell numbers recovered within 3 days of α-GalCer administration, indicating repopulation of iNKT cells after stimulation with α-GalCer (20, 21). However, the role of iNKT cell repopulation during the late phase of hepatic I/R injury is poorly understood. In the present study, we investigated the correlation between the repopulation of hepatic iNKT cells and liver repair after hepatic I/R injury in mice.
Materials and Methods
Animals. Male C57BL/6 mice (8-9 weeks old) were purchased from CLEA Japan (Tokyo, Japan). All mice were housed in specific pathogen-free conditions with free access to water and food in a 12-h light/dark cycle. All experimental procedures were conducted in accordance with the protocol approved by the Animal Experimentation and Ethics Committee of the Kitasato University School of Medicine (2021-091).
Animal procedures. Hepatic I/R injury was induced as previously described (16). Briefly, mice were anesthetized with an intraperitoneal (i.p.) injection of a combination cocktail containing medetomidine hydrochloride (0.3 mg/kg, Nippon Zenyaku Kogyo, Fukushima, Japan), midazolam (4.0 mg/kg, Astellas Pharma, Tokyo, Japan), and butorphanol (5.0 mg/kg, Meiji Seika Pharma, Tokyo, Japan). Hepatic ischemia was performed using vascular clamps (ROBOZ Surgical Instrument, Washington, DC, USA) to interrupt the blood supply to the median and left hepatic lobes. After 60 min of partial hepatic ischemia, reperfusion was initiated by removing the clamps. Medetomidine effects were reversed with the i.p. injection of atipamezole (0.75 mg/kg, Nippon Zenyaku Kogyo). Sham control mice underwent the same protocol without vascular occlusion. For the analyses of blood, histology, and mRNA, 6 animals were used in the vehicle-treated sham group, 5 animals in the α-GalCer-treated sham group, 6 animals in the vehicle-treated I/R group, and 6 animals in the α-GalCer-treated I/R group. For flow cytometry analysis, 4 animals were used in each group.
Experimental protocols. Animals were i.p. injected with α-GalCer (0.1 mg/kg body weight, Funakoshi, Tokyo, Japan) dissolved in 0.1% DMSO and diluted in phosphate-buffered saline (PBS) or PBS containing 0.1% DMSO (vehicle) at the induction of ischemia (16). Mice were euthanized with isoflurane after the indicated reperfusion periods. Blood was taken from the heart. Serum alanine aminotransferase (ALT) activity was measured using a Dri-Chem 7000 Chemistry Analyzer System (Fujifilm, Tokyo, Japan). Immediately after blood collection, livers were excised. A small section of each liver was placed in 10% formaldehyde and the remaining liver tissue was immersed in RNAiso Plus (Takara Bio, Shiga, Japan) for isolating RNA to perform polymerase chain reaction (PCR) quantification.
Histology and immunohistochemistry. Paraffin sections of the liver tissues were prepared for either hematoxylin and eosin (H&E) staining or immunostaining. A microscope (Biozero BZ-700 Series; Keyence, Osaka, Japan) was used to capture images of H&E- or immune-stained sections. The level of hepatic necrosis was estimated by measuring the necrotic area in H&E-stained sections. Sections were also stained using an antibody against proliferating cell nuclear antigen (PCNA) (Thermo Fisher Scientific, Waltham, MA, USA). Immune complexes were detected with Histofine Simple Stain MAX PO (MULTI) (Nichirei, Tokyo, Japan). The number of PCNA+ hepatocytes was counted in five fields (400×) per animal. The results are expressed as percentages of PCNA+ hepatocytes.
Quantitative real-time RT-PCR analysis. Total liver RNA was extracted from tissues using RNAiso Plus (Takara Bio). Single-stranded cDNA was generated from 1 μg of total RNA by reverse transcription using ReverTra Ace qPCR RT kits (Toyobo, Osaka, Japan). Quantitative PCR was performed using TB Green Premix Ex Taq II (Tli RNaseH Plus; Takara Bio, Shiga, Japan). Primer sequences used in the present study are listed in Table I. Data were normalized to the expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in each sample.
The primers used for reverse transcription and quantitative polymerase chain reaction (PCR).
Isolation of intrahepatic leukocytes. Under anesthesia, the liver was perfused with Hank’s balanced salt solution through the portal vein. Excised livers were incubated in RPMI containing 0.05% collagenase (Type IV; Sigma Chemical Co., St. Louis, MO, USA) for 20 min at 37°C. The liver homogenates were filtered through a 70 μm cell strainer. Non-parenchymal cells were purified by density-gradient centrifugation on 33% Percoll (GE Healthcare Life Sciences, Piscataway, NJ, USA) as previously reported (16).
Flow cytometric analyses. Isolated non-parenchymal cells were incubated with anti-mouse CD16/32 antibody (BioLegend, San Diego, CA, USA) to block non-specific binding of primary mAb. Then, the cells were stained with a combination of the following reagents; PE-conjugated anti-CD45 (30-F11, BioLegend), APC/CY7-conjugated anti-Ly6G (1A8, BioLegend), PE/Cy7-conjugated anti-CD11b (M1/70, BioLegend), Brilliant Violet 510-conjugated anti-Ly6C (HK1.4, BioLegend), anti-F4/80 (BM8, BioLegend), FITC-conjugated anti-TCRβ (H57-597, BioLegend), and α-GalCer (PBS-57)-loaded CD1d tetramer. Control CD1d-tetramer was kindly provided by The NIH Tetramer Core Facility at Emory University (Atlanta, GE, USA). Cells positive for 7-aminoactinomycin D (BioLegend) were excluded from the analysis. Samples were analyzed on a FACS Verse cytometer (BD Biosciences, Franklin Lakes, NJ, USA). Data were analyzed using Kaluza software v2.1 (Beckman Coulter, Brea, CA, USA). We quantified and presented the numbers of cells normalized to per liver tissue weight (no. cells/g).
For intracellular cytokine staining, cells were fixed and permeabilized using the FOXP3 Fix/Perm Buffer Set (BioLegend) after surface marker staining. Treated cells were stained with APC-conjugated anti-IFN-γ mAb (XMG1.2, BioLegend), APC-conjugated anti-IL-4 mAb (11B11, BioLegend), and PE/Cy7 conjugated anti-IL-13 mAb (eBio 13A, eBioscience, San Diego, CA, USA). Cells were analyzed by flow cytometry (FACS Verse cytometer, BD Biosciences) (16).
For cell proliferation, mice were administered 5 mg/kg 5-ethynyl-2’-deoxyuridine (EdU, Invitrogen, Carlsbad, CA, USA) by i.p. injection 3 h before being sacrificed. Cells were measured using Click-iT EdU flow cytometry assay kits (Invitrogen).
Statistical analyses. Results are presented as mean±standard deviation (SD). Data were analyzed using GraphPad Prism software, version 8 (GraphPad Software, La Jolla, CA, USA). Data were compared between multiple groups using one-way analyses of variance followed by Tukey’s post-hoc tests. All p-values <0.05 were considered statistically significant. Significance is represented with asterisks as following: *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
Results
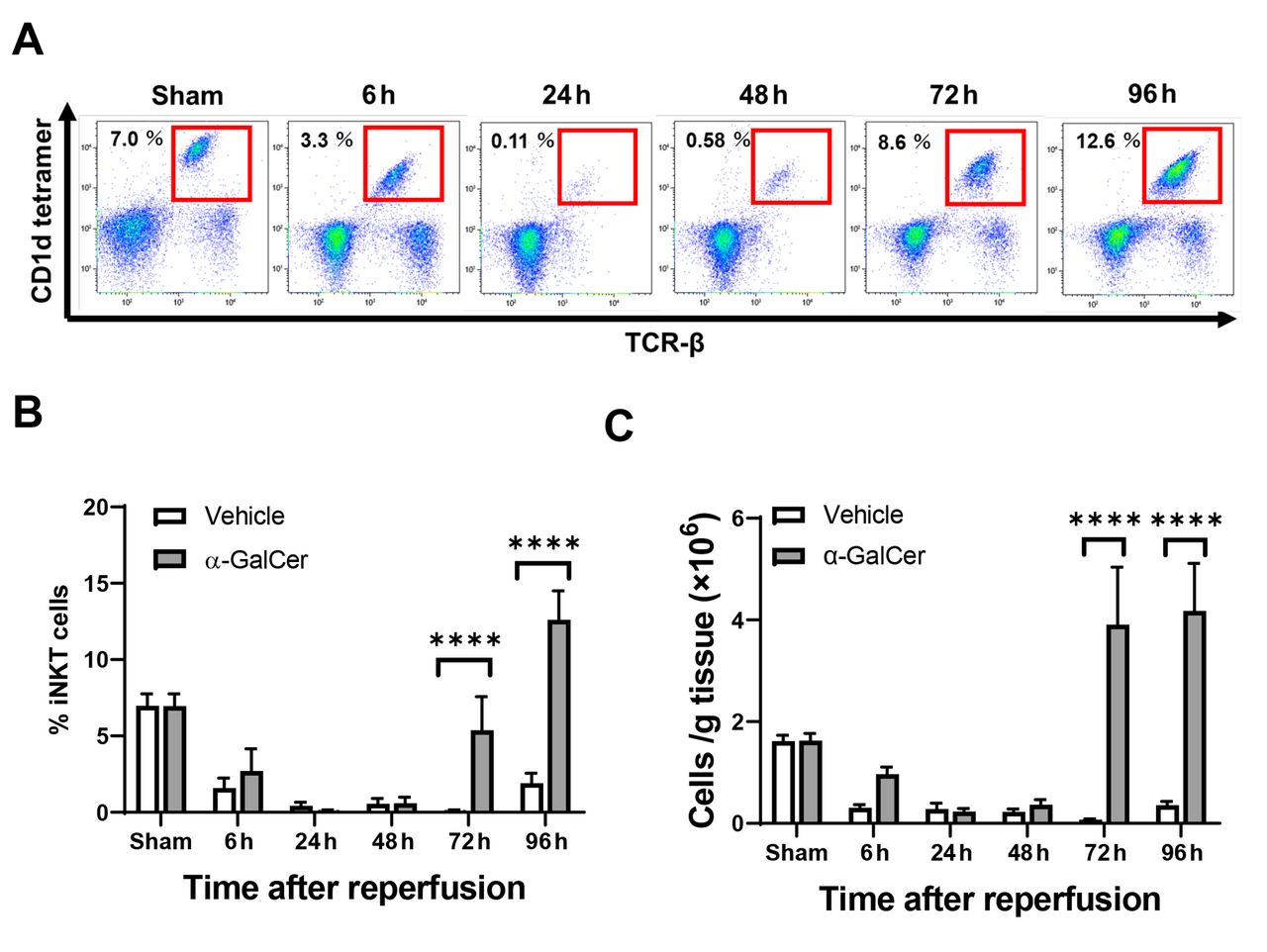
Dynamics of iNKT cell populations after hepatic I/R. We first confirmed the dynamics of iNKT cells during hepatic I/R (Figure 1). When iNKT cells were identified as TCRβ+ and CD1d-tetramer+ using flow cytometry analysis (Figure 1A), numbers of iNKT cells in α-GalCer-treated mice decreased from 6 h post-reperfusion to 48 h post-reperfusion (Figure 1B, C). These results indicate that the expression of cell surface markers for iNKT cells was down-regulated, which prevents visualization of iNKT cells by flow cytometric detection (21). Numbers of iNKT cells in α-GalCer-treated mice then rapidly and markedly increased to levels substantially higher than in the sham group at 96 h post-reperfusion. In contrast, numbers of iNKT cells in vehicle-treated mice decreased from 6 h post-reperfusion and remained low until up to 96 h post-reperfusion.
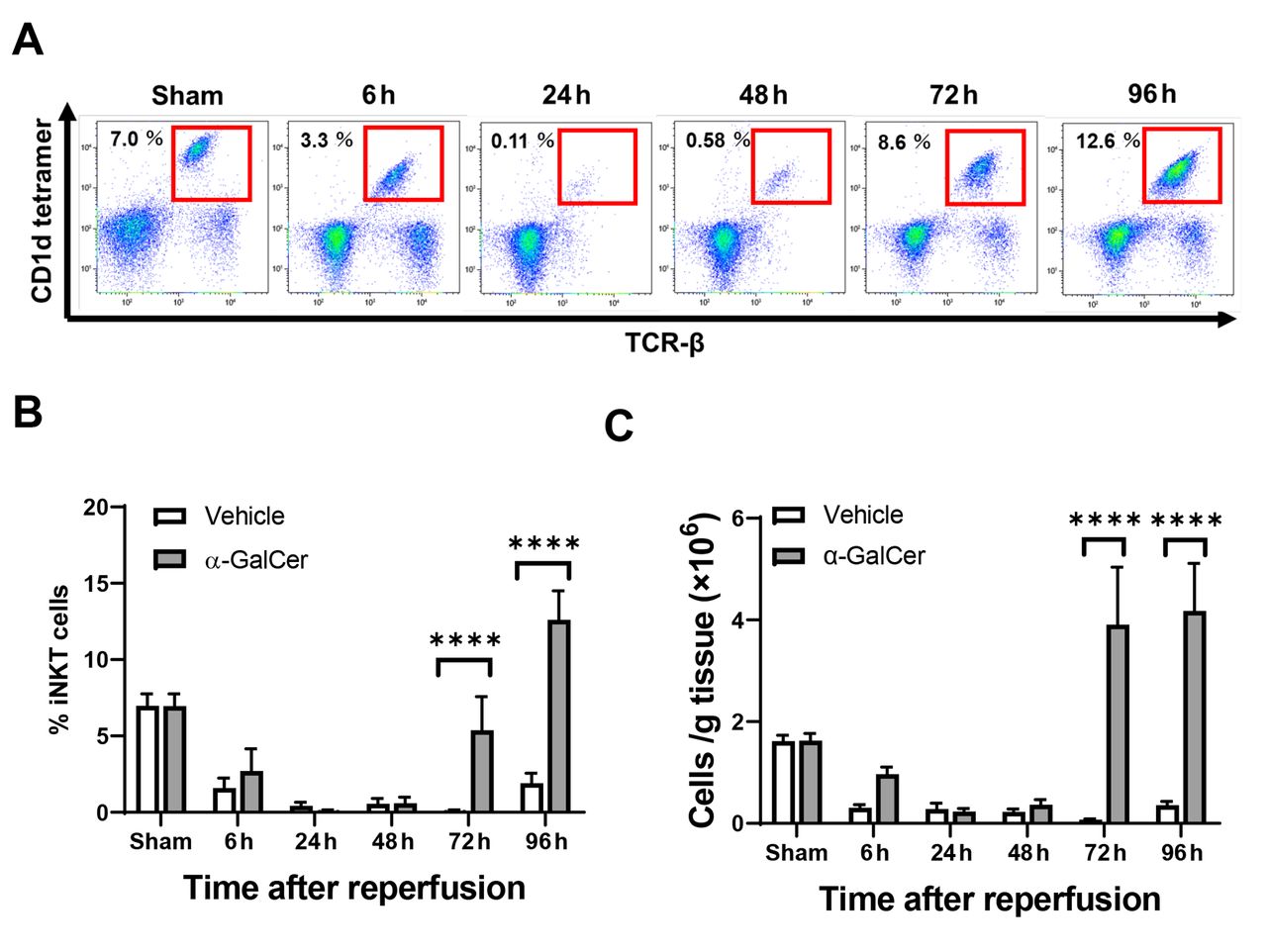
Changes in hepatic invariant natural killer T (iNKT) cells in mice treated with α-galactosylceramide (α-GalCer) or vehicle after hepatic ischemia reperfusion (I/R). (A) Representative dot plots of iNKT cells in mice treated with α-GalCer during hepatic I/R. (B) The percentages of hepatic iNKT cells out of CD45+ cells in mice treated with α-GalCer or vehicle after hepatic I/R. (C) The numbers of hepatic iNKT cells in mice treated with α-GalCer or vehicle after hepatic I/R. Data are expressed as the mean±SD (n=4 mice per group). ****p<0.0001.
Repopulation of iNKT cells is associated with liver repair after hepatic I/R injury. We also determined the numbers of iNKT cells in α-GalCer-treated mice at 96 h after sham operation. As shown in Figure 2A, the number of iNKT cells in α-GalCer-treated livers of the sham group was higher than those in the vehicle-treated sham group. Treatment with α-GalCer further increased the number of hepatic iNKT cells in the hepatic I/R group compared to the sham group. No statistically significant difference in ALT levels was observed between mice treated with α-GalCer and vehicle at 96 h post-reperfusion (Figure 2B). However, treatment with α-GalCer reduced hepatic necrosis at 96 h post-reperfusion compared to vehicle-treated mice (Figure 2C). In addition, the expression of PCNA, a marker of hepatocyte proliferation, was higher in mice treated with α-GalCer than that in mice treated with vehicle (Figure 2D). These results indicate that α-GalCer administration in mice with hepatic I/R induced re-expansion of iNKT cells, which was associated with liver repair and resolution of liver inflammation as demonstrated by reduced liver necrosis and upregulation of PCNA expression. We further examined the effect of α-GalCer administration alone on liver inflammation and repair. No necrosis was observed in mice treated with α-GalCer or vehicle after sham operation (Figure 2C). There was a slight increase in PCNA expression after sham operation in α-GalCer-treated mice compared to vehicle-treated mice; however, this difference did not reach statistical significance (Figure 2D). Thus, these results demonstrate that α-GalCer administration in mice following a sham operation induced re-expansion of iNKT cells, although to a lesser extent than in mice with hepatic I/R, indicating the effects of α-GalCer on iNKT cell numbers may occur also in the absence of liver injury.

Reappearance of hepatic invariant natural killer T (iNKT) cells is associated with liver repair after hepatic ischemia reperfusion (I/R) injury. (A) Numbers of hepatic iNKT cells in mice treated with α-galactosylceramide (α-GalCer) or vehicle at 96 h after hepatic I/R or sham. Data are expressed as the mean±SD (n=4 mice per group). *p<0.05, ****p<0.0001. (B) Alanine aminotransferase (ALT) levels in mice treated with α-GalCer or vehicle at 96 h after hepatic I/R or sham. Data are expressed as the mean±SD (n=5-6 mice per group). (C) Hepatic necrosis in mice treated with α-GalCer or vehicle at 96 h after hepatic I/R or sham. Data are expressed as the mean±SD (n=5-6 mice per group). ****p<0.0001. Representative hematoxylin and eosin (H&E)-stained liver images from mice treated with α-GalCer or vehicle at 96 h after hepatic I/R or sham. Scale bar, 200 μm. (D) Representative immunohistochemical images of staining for proliferating cell nuclear antigen (PCNA) in livers of mice treated with α-GalCer or vehicle at 96 h after hepatic I/R or sham. Arrows indicate PCNA+ hepatocytes. Scale bar, 100 μm. Bar chart shows the quantitative analysis of the corresponding staining. Data are expressed as the mean±SD (n=4-5 mice per group). *p<0.05, ****p<0.0001.
Functional changes in iNKT cells in the late phase of hepatic I/R. To investigate whether the expansion of iNKT cells in livers at 96 h post-reperfusion was due to proliferation, we measured the expression of EdU, a marker of cell proliferation, in iNKT cells (Figure 3A). α-GalCer increased the numbers of EdU-positive cells in the livers of mice following sham surgery and hepatic I/R, with approximately 50% of expanded iNKT cells positive for EdU. However, no difference in the numbers of EdU-positive iNKT cells following α-GalCer administration was observed between the sham and hepatic I/R groups. These results suggest that α-GalCer administration induced expansion of iNKT cells partly through local proliferation of iNKT cells. As activated iNKT cells produce a mixture of T helper type 1 (Th1) and Th2 cytokines such as IFN-γ, interleukin (IL)-4, and IL-13, we measured the levels of these cytokines in iNKT cells at 96 h post-reperfusion (Figure 3B). There was no statistical difference in IFN-γ levels in hepatic iNKT cells at 96 h post-reperfusion between mice treated with α-GalCer and vehicle; however, IFN-γ levels in hepatic iNKT cells after α-GalCer administration were higher in the sham group compared to the hepatic I/R group. On the other hand, IL-13 levels in hepatic iNKT cells at 96 h post-reperfusion from mice treated with α-GalCer were higher than those from mice treated with vehicle. In addition, IL-13 levels in iNKT cells following α-GalCer administration were higher in the hepatic I/R group compared to the sham group. No difference in IL-4 levels in iNKT cells at 96 h post-reperfusion were observed between mice treated with α-GalCer and vehicle. These results suggest that proliferating iNKT cells induced by α-GalCer produced mainly IL-13 at 96 h post-reperfusion.

Proliferation of invariant natural killer T (iNKT) cells and production of cytokines from iNKT cells after hepatic ischemia reperfusion (I/R). (A) 5-Ethynyl-2’-deoxyuridine (EdU)-positive iNKT cells in mice treated with α-galactosylceramide (α-GalCer) or vehicle at 96 h after hepatic I/R or sham. Data are expressed as the mean±SD (n=3-4 mice per group). *p<0.05. (B) Proportion of interferon (IFN)-γ-, interleukin (IL)-13-, and IL-4-positive iNKT cells in mice treated with α-GalCer or vehicle at 96 h after hepatic I/R or sham. Data are expressed as the mean±SD (n=4 mice per group). *p<0.05.
Accumulation of macrophages in the repair phase of hepatic I/R. Increased expression of genes related to pro-inflammatory macrophages and reparative macrophages occurs in liver tissues during the repair phase of hepatic I/R injury (3,4). Real-time PCR analyses demonstrated that the levels of mRNA encoding pro-inflammatory macrophage-related genes [nitric oxide synthetase-2 (Nos2)] and mRNA encoding reparative macrophage-related genes [resistin-like molecule-α, Retnla, and interleukin-10 (I110)] at 96 h post-reperfusion were higher in mice treated with α-GalCer than in mice treated with vehicle (Figure 4A, B). In addition, in mice treated with α-GalCer, we observed increased expression of mRNA encoding pro-inflammatory macrophage-related genes [interleukin-6 (Il6) and Nos2] and mRNA encoding reparative macrophage-related genes (Retnla and I110) in the I/R group compared to the sham group, at 96 h post-reperfusion (Figure 4A, B).

Infiltration of hepatic macrophages in the late phase of hepatic ischemia reperfusion (I/R) injury. (A) Expression of genes related to pro-inflammatory macrophage phenotypes [interleukin (Il)6 and nitric oxide synthetase (Nos)2] and (B) genes related to reparative macrophage phenotypes [resistin-like molecule-α (Retnla) and Il10] in mice treated with α-galactosylceramide (α-GalCer) or vehicle at 96 h after hepatic I/R or sham. Data are expressed as the mean±SD (n=4-5 mice per group). **p<0.01, ***p<0.001, ****p<0.0001. (C) Flow cytometry gating strategy of liver macrophages in mice treated with α-GalCer at 96 h after hepatic I/R. Lymphocyte antigen 6 complex, locus G (Ly6G)−/Ly6Chigh/CD11bhigh/F4/80high cells were defined as pro-inflammatory macrophages (Ly6Chigh macrophages) and Ly6G−/Ly6Clow/CD11bhigh/F4/80high cells were defined as reparative macrophages (Ly6Clow macrophages). (D) Changes in the numbers of pro-inflammatory and reparative macrophages in livers from mice treated with α-GalCer or vehicle after hepatic I/R injury or sham operation. Data are expressed as the mean±SD (n=4-6 mice per group). ****p<0.0001.
These results indicate that α-GalCer treatment increased the hepatic expression of genes related to pro-inflammatory and reparative macrophages at 96 h post-reperfusion. We further measured hepatic accumulation of macrophages during the late phase of hepatic I/R by flow cytometry analysis. At 96 h post-reperfusion, the numbers of pro-inflammatory macrophages, namely Ly6G−/Ly6Chigh/CD11bhigh/F4/80high cells (Ly6Chigh macrophages), were higher in α-GalCer-treated mice than in vehicle-treated mice, in both I/R and sham groups (Figure 4C). However, no difference in the number of pro-inflammatory macrophages after α-GalCer administration was observed between the hepatic I/R and sham groups. Additionally, the numbers of reparative macrophages, namely the Ly6G−/Ly6Clow/CD11bhigh/F4/80high cells (Ly6Clow macrophages), were higher in α-GalCer-treated compared to vehicle-treated mice in the I/R group (Figure 4C), while in the sham group, there was no statistical difference between mice treated with α-GalCer and vehicle. Furthermore, higher numbers of Ly6Clow macrophages following α-GalCer administration were observed in the hepatic I/R group compared to the sham group. Taken together, α-GalCer administration induced hepatic iNKT cell repopulation and increased accumulation of Ly6Clow macrophages at 96 h post-reperfusion.
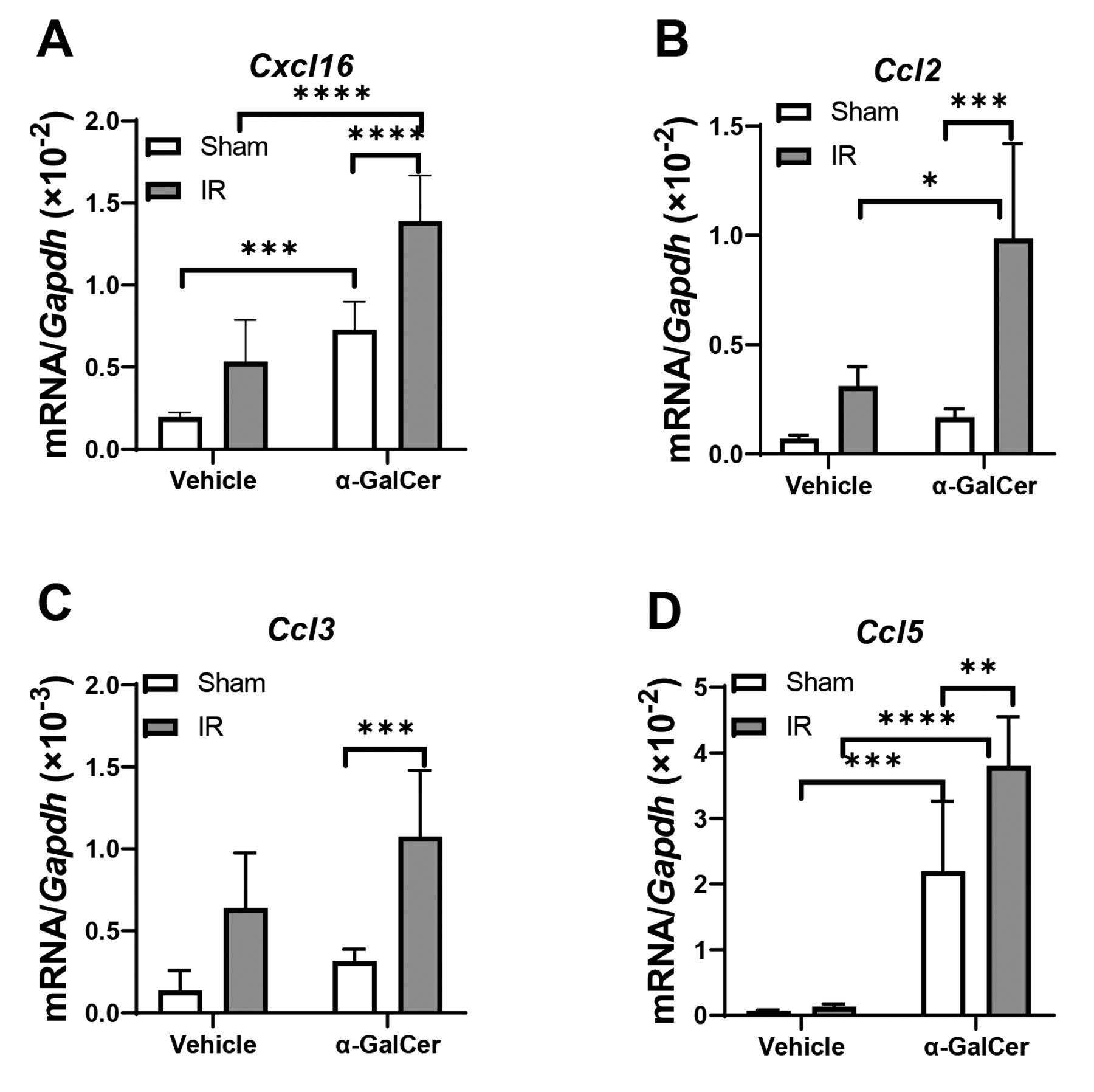
In line with increases in the numbers of hepatic iNKT cells and macrophages, we examined the expression levels of mRNA encoding Cxcl16, which is a chemokine for recruitment of iNKT cells (2) (Figure 5A). α-GalCer treatment increased mRNA levels of Cxcl16 in livers from mice in the sham and hepatic I/R groups compared to vehicle treatment. Hepatic mRNA levels of Cxcl16 in α-GalCer-treated mice were higher in the hepatic I/R group compared to the sham group. Regarding chemokines involved in the recruitment of macrophages, we found that hepatic expression levels of mRNA encoding Ccl2 and Ccl5 but not Ccl3 were higher at 96 h post-reperfusion in α-GalCer-treated mice compared to vehicle-treated mice, in the I/R group (Figure 5B, C, D). Higher levels of Ccl2, Ccl3, and Ccl5 mRNA expression following α-GalCer administration were observed in the hepatic I/R group compared to the sham group. These results indicate that upregulation of the chemokines CXCL16, CCL2, CCL3 and CCL5 contributes to α-GalCer-induced hepatic accumulation of iNKT cells and macrophages at 96 h post-reperfusion.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cytokine expression levels after hepatic ischemia reperfusion (I/R). mRNA levels of C-X-C motif chemokine ligand (Cxcl)16 (A), C-C motif chemokine (Ccl)2 (B), Ccl3 (C), and Ccl5 (D) in livers from mice treated with α-galactosylceramide (α-GalCer) or vehicle at 96 h after hepatic I/R injury or sham operation. Data are expressed as the mean±SD (n=4-6 mice per group). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Discussion
The objective of the present study was to examine whether expansion of hepatic iNKT cells during the late phase of hepatic I/R injury contributes to liver repair. We demonstrated that repopulation of hepatic iNKT cells stimulated with α-GalCer was associated with enhancement of liver repair. In addition, proliferated iNKT cells in the late phase of hepatic I/R appeared to predominantly produce IL-13, which was accompanied by the accumulation of reparative macrophages. These results indicate that the expansion of iNKT cells contributes to liver repair following hepatic I/R injury by promoting the hepatic accumulation of macrophages.
Previous studies have shown conflicting results including protective or deleterious roles of iNKT cells in the early phase of hepatic I/R injury (11-13). However, little is known about the involvement of iNKT cells in liver repair after acute liver injury. Recent studies indicate that iNKT cells contribute to the resolution of inflammation and tissue repair after focal thermal trauma in the liver by polarization of monocyte-derived macrophages accumulated in the injured regions (15). In addition, we have reported that activated iNKT cells stimulated with α-GalCer facilitate liver recovery through acceleration of macrophage phenotypic switching from pro-inflammatory to reparative phenotypes in the early phase of hepatic I/R injury (16). These findings suggest that iNKT cells stimulate liver tissue recovery during the early stages of acute liver injury. On the other hand, our data showed that repopulated iNKT cells after administration of α-GalCer contribute to liver repair in the late phase of hepatic I/R injury by promoting the accumulation of reparative macrophages.
α-GalCer administration is known to cause rapid reduction in the numbers of hepatic iNKT cells followed by the reappearance of iNKT cells within 3 days (19, 20). Consistent with this, the administration of α-GalCer was found to increase the number of hepatic iNKT cells at 96 h post-reperfusion in the present study, with higher numbers of hepatic iNKT cells observed in mice with hepatic I/R compared to mice that underwent the sham operation. In addition, hepatic mRNA levels of Cxcl16, a chemokine involved in the recruitment of iNKT cells, were higher in α-GalCer-treated mice with hepatic I/R than in α-GalCer-treated mice that underwent the sham operation. Thus, increased levels of chemokines in response to hepatic I/R injury may induce expansion of hepatic iNKT cells. As activated iNKT cells at 96 h post-reperfusion were positive for EdU, the reappearance of hepatic iNKT cells may be due to local proliferation of iNKT cells.
Compared to vehicle-treated mice, α-GalCer-treated mice had reduced hepatic necrosis and increased PCNA expression at 96 h post-reperfusion, indicating that expanded iNKT cells are involved in the promotion of liver repair after hepatic I/R injury. Further, we observed increased levels of genes related to pro-inflammatory macrophages, including Il-6 and Nos2, and reparative macrophages, including Retnla and Il-10. In addition, the numbers of pro-inflammatory and reparative macrophages were higher in α-GalCer-treated mice than in vehicle-treated mice at 96 h post-reperfusion. As levels of chemokines including CCL2 and CCL5 were higher in α-GalCer-treated mice than in vehicle-treated mice at 96 h post-reperfusion, macrophages were seen to accumulate in areas of tissue injury in response to upregulation of these chemokines. These results indicate that the expansion of iNKT cells facilitated liver repair after hepatic I/R injury through the recruitment and accumulation of macrophages.
The reappearance of hepatic iNKT cells was observed after administration of α-GalCer in the I/R and sham groups. When compared with iNKT cells isolated from mice that had undergone the sham operation, iNKT cells from mice with hepatic I/R had increased levels of IL-13 but not IL-4 or IFN-γ. These results indicate that iNKT cells from mice with hepatic I/R mediated macrophage phenotypic switching through the production of IL-13 in the late phase of hepatic I/R injury. By contrast, iNKT cells isolated from mice that had undergone the sham operation produced IFN-γ but not IL-4 or IL-13. These results corroborate a previous study reporting that iNKT cells continue to produce IFN-γ but not IL-4 for at least 72 h after the administration of α-GalCer (20). These findings suggest that iNKT cells produced IL-13 in response to liver injury during the late phase of hepatic I/R. CD1d is essential for presentation of α-GalCer to iNKT cells (6). We have previously shown that pro-inflammatory and reparative macrophages express CD1d to interact with activated iNKT cells during the early phase of hepatic I/R (16). These findings suggest that activated iNKT cells may interact with CD1d-expressing macrophages accumulating in areas of tissue injury during the late phase of hepatic I/R to produce IL-13, thereby stimulating macrophage polarization (4, 22). Further studies are required to clarify the role of IL-13 produced from iNKT cells in liver repair after hepatic I/R injury.
Accumulating evidence indicates that iNKT cells play a crucial role in early immune responses by producing large amounts of Th1- and Th2-associated cytokines, including IFN-γ, IL-4, and IL-13, resulting in macrophage activation (17, 20). Activation of iNKT cells by α-GalCer leads to rapid production of IL-13 to protect the liver against hepatic I/R injury (13). IL-4 derived from iNKT cells at 6 h post-reperfusion promotes liver repair after hepatic I/R injury (16). These results indicate that iNKT cells are involved in rapid responses to stimuli by the innate immune system. In addition, the results of the present study indicate that the reappearance of iNKT cells also contributes to the production of cytokines related to macrophage polarization through iNKT cell-macrophage crosstalk during the repair phase of hepatic I/R injury (17).
The present study has several limitations. First, we demonstrated that expansion of iNKT cells during the late phase of hepatic I/R injury was associated liver repair, though additional studies using adoptive transfer experiments of repopulated iNKT cells into mice or NKT cell-deficient mice will be needed to confirm the involvement of repopulated iNKT cells. Second, we provided the obtained results in hepatic I/R injury model. To further understand the role of the expanded iNKT cells in liver repair after acute liver injury, other liver disease models should be explored. Moreover, the results of the current study do not contain any data from patients.
In conclusion, expansion of iNKT cells stimulated with α-GalCer following hepatic I/R injury promotes the accumulation of macrophages in regions of tissue injury to facilitate liver repair. α-GalCer administration promotes liver repair after hepatic I/R injury by promoting the expansion of iNKT cells and macrophages. Activation of iNKT cells by α-GalCer may serve as a good option for the treatment of liver repair and regeneration after acute liver injury.
Acknowledgements
The Authors thank Michiko Ogino and Kyoko Yoshikawa for their technical assistance and the NIH Tetramer Core Facility for the control and ligand-loaded mouse CD1d-tetramer. This work was supported by Grants from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (MEXT) (20K17630 to NN and 22K08856 to YI).
Footnotes
Authors’ Contributions
TG conceived the project, carried out the experiments, performed data collection, data analysis, and wrote the manuscript. YI reviewed pathological data, analyzed the data, and wrote the manuscript. YK, SN, NN, and KH performed the experiments and analyzed the data. TN, NH, and HA designed the research, supervised the project, and reviewed the manuscript.
Conflicts of Interest
The Authors have no conflicts of interest to declare for this study.
- Received September 6, 2022.
- Revision received October 7, 2022.
- Accepted October 13, 2022.
- Copyright © 2022, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY-NC-ND) 4.0 international license (https://creativecommons.org/licenses/by-nc-nd/4.0).