

Abstract
Background/Aim: The combination of regorafenib with cisplatin/pemetrexed has indicated controllable safety and encouraging antitumor activity in non-small cell lung cancer (NSCLC) patients. However, the anti-NSCLC effects and action mechanisms of regorafenib combined with cisplatin is ambiguous. The major goal of the study was to study the inhibitory effects and action mechanisms of regorafenib combined with cisplatin in NSCLC cells. Materials and Methods: Cell viability, flow cytometry, immunofluorescence staining, western blotting, migration, and invasion assays were employed to verify the anti-NSCLC effects and mechanisms of regorafenib in combination with cisplatin. Results: Cisplatin-induced epidermal growth factor receptor (EGFR)/nuclear factor κB (NF-κB) signaling was effectively inhibited by regorafenib treatment. Regorafenib, erlotinib (EGFR inhibitor) and QNZ (NF-κB inhibitor) may all enhance the cytotoxicity effect of cisplatin. The invasion ability was effectively decreased by combination treatment. Caspase-dependent and -independent apoptosis was activated by cisplatin combined with regorafenib. Conclusion: Apoptosis induction and EGFR/NF-κB inactivation correlate with regorafenib-enhanced anti-NSCLC efficacy of cisplatin. This study provides evidence of the therapeutic efficacy of regorafenib in combination with cisplatin on NSCLC.
- Regorafenib
- cisplatin
- EGFR
- NF-κB
- non-small cell lung cancer
Cisplatin, a platinum-based anticancer drug, is standard treatment for advanced non-small cell lung cancer (NSCLC). DNA damage-induced apoptosis correlates with the anticancer mechanism of cisplatin (1, 2). Cell and animal models have revealed that enhancement of apoptosis induction and suppression of oncogenic signaling such as Wnt, AKT, epidermal growth factor receptor (EGFR), and nuclear factor-kappaB (NF-κB) by potential complementary approaches have been reported to sensitize NSCLC cells to cisplatin (3-6). For instance, sodium valproate, a histone deacetylase inhibitor, and microRNA miR-381 have been found to enhance anti-NSCLC efficacy of cisplatin through inducing apoptosis and inhibiting NF-κB signaling, respectively (7, 8).
Several tyrosine kinase inhibitors (TKIs) targeting EGFR and anaplastic lymphoma kinase (ALK) have been shown to improve survival of NSCLC patients receiving platinum-based doublet chemotherapy (9, 10). Regorafenib, an oral multi-target TKI, is used for the treatment of solid tumors such as hepatocellular carcinoma (HCC), renal cell carcinoma, differentiated thyroid cancer, and metastatic colorectal cancer (11, 12). Induction of apoptosis and down-regulation of AKT/NF-κB signaling were associated with regorafenib-inhibited progression of NSCLC cells (13). In addition, the combination of regorafenib with cisplatin/pemetrexed has shown manageable tolerability and encouraging antitumor activity in NSCLC patients (14). However, the anticancer effect and action mechanism of regorafenib combined with cisplatin has not yet been elucidated. The main purpose of the study was to verify anti-proliferative, anti-invasive, and apoptotic effects and mechanisms of regorafenib in combination with cisplatin in NSCLC cells.
Materials and Methods
Cell lines. Human lung adenocarcinoma cell line, A549 was maintained in RPMI-1640 medium (Thermo Fisher Scientific, Fremont, CA, USA) with 10% fetal bovine serum (FBS) 2 mM L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin (Thermo Fisher Scientific), at a 37°C incubator with 5% CO2 and 95% humidity.
Reagents and antibodies. The chemical reagents purchased from Sigma (St.Louis, MO, USA) were listed as follows: 3-(4, 5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT); QNZ (NF-κB inhibitor); rgorafenib, cisplatin and dimethyl sulfoxide (DMSO). Primary antibodies against MMP-9, phosphor-NF-κB ser536, NF-κB, Cyclin D1, VEGF-A, β-actin, α-tubulin were obtained from Elabsicence (Houston,TX USA). Primary antibodies against MMP-2 (proteintech, Rosemont, IL, USA) and phosphor-EGFR Tyr1068 (Cell Signaling, Danvers, MA, USA) were purchased as indicated.
Cell viability assay. A549 cells were seeded in 96-well plates, at a density of 5×103 cells/well overnight and treated with regorafenib 2.5-30 μM, cisplatin 2.5-30 μM and regorafenib 10 μM combine different concentrations (5~20 μM) of cisplatin for 48h. A549 cells were also treated with 0-5 μM QNZ and combined with 5-20 μM cisplatin for 48 h. The medium was then replaced by 100 μl MTT reagent (0.5 mg/ml) and maintained for another 2h. Finally, the MTT medium were be removed and replaced with 100 μl DMSO for further dectection. The absorbance was dectected by SpectraMax iD3 microplate reader (Molecular Devices, San Jose, CA, USA) at 570 nm (15).
Caspase-3, -8, -9 activity analyses. A549 cells were seeded in 6 well plates, at a density of 1×105 cells/well overnight and treated with regorafenib 10 μM and/or cisplatin 15 μM for 48 h. A549 cells were further stained with active caspase-3, caspase-8 and caspase-9 staining Kit (CaspGLOW™ fluorescein staining kit, BioVision) for 30 min at 37°C incubator. The activate caspase-3 and caspase-9 were determined by FL-1 channel. The activate caspase-8 was determined by FL-2 channel using NovoCyte flow cytometry with NovoExpress® software (Agilent Technologies Inc., Santa Clara, CA, USA). The quantification was performed by FlowJo software (16).
Fas activity analyses. A549 cells were seeded in 6 well plates, at a density of 1×105 cells/well overnight and treatment regorafenib 10 μM and/or cisplatin 15 μM for 48 h. A549 cells were further stained with FITC conjugated Fas (Thermo Fisher Scientific) in 100 μl binding buffer for 15 min in the dark at room temperature (17). The Fas was determined by FL-1 channel using NovoCyte flow cytometry with NovoExpress® software (Agilent Technologies Inc. and quantified by FlowJo software.
Invasion and migration assay. Transwell chambers (BD Biosciences, Franklin Lakes, NJ, USA) were coated with (migration) or without (invasion) 50 μl matrigel in serum free medium (1:1) overnight. A549 cells were plated in 10 cm and seeding with 1×106 cells/plate followed with treatment of regorafenib 10 μM and/or cisplatin 15 μM for 48 h. The viable A549 cells (2×105) were re-suspended in 200 μl serum-free medium and added to the upper chamber. The 10% FBS/RPMI medium were placed into the lower chamber of cell migration/invasion inserts. Cells were allowed for migration or invasion for 48 h, then cells were be fixed with 4% paraformaldehyde in PBS for 30 min at 4°C, stained with crystal violet solution for 15 min and photographed by microscope (Nikon ECLIPSE Ti-U, Minato City, Tokyo, Japan), and quantified by ImageJ software version 1.50 (National Institutes of Health, Bethesda, MD, USA) (18, 19).
Western blotting assay. A549 cells were plated in 10 cm, seeded with 1×106 cells and treated with regorafenib 10 μM, cisplatin 15 μM and combination for 48 h. After treatment, A549 cells were collected and extracted with total proteins by NP-40 lysis buffer containing proteinase inhibitor cocktail and phosphatase inhibitor (Sigma-Aldrich). The protein concentration was measured by Bradford method (Bio-Rad). Forty microgram of protein per group were separated by 10-15% SDS-page and transferred onto polyvinylidene difluoride (PVDF) membranes (EMD Millipore, Bedford, MA, USA). Membranes were blocked with blocking buffer (5% non-fat dry milk), hybridized with the first antibody and reactivated with horseradish peroxidase (HRP)-conjugated second antibody. Chemiluminescence images were detected by the chemiluminescent image system (ChemiDoc-It 515, UVP, Upland, CA, USA) (20).
Immunofluorescence staining of AIF. A549 cells were seeded onto 4-well Nunc™ Lab-Tek™ II Chamber slide™ (Thermo Fisher Scientific, Fremont, CA, USA) overnight at 2.5×104 cells/well density. A549 cells were treated with regorafenib 10 μM, cisplatin 15 μM and combination for 48 h. Cells were then fixed, permeabilized by 4% paraformaldehyde, blocked by 1% BSA, stained by AIF and followed with Alexa Fluor 488 secondary antibody conjugation. Detail procedure was described in previous study (21). AIF nuclear translocation was finally imaged by Zeiss Axio Scope.A1 fluorescence microscope (Germany).
Statistical analysis. Statistical significance was measured by one-way ANOVA analysis. p-values<0.05, <0.01 and <0.005 were all considered as statistically significant. The statistical analysis was conducted using the GraphPad Prism 7. All experiments were independently repeated at least three times.
Results
Regorafenib-enhanced cytotoxicity of cisplatin is associated with EGFR/NF-κB inactivation in NSCLC. In Figure 1A and B, the viability of A549 cells was decreased by regorafenib and cisplatin alone in a dose-dependent manner. As illustrated in Figure 1C, the combination of regorafenib with cisplatin effectively increased cytotoxicity in A549 cells. To identify the action mechanism of regorafenib combined with cisplatin, NF-κB inhibitor and EGFR inhibitor (erlotinib) combined with cisplatin were used to evaluate cytotoxicity on A549 cells. In Figure 1D and E, cell viability was also reduced by QNZ alone and combined with cisplatin. In addition, the cytotoxicity of cisplatin was also triggered by erlotinib (Figure 1F). Moreover, regorafenib significantly suppressed cisplatin-induced phosphorylation of EGFR and NF-κB. In sum, the toxicity induction of cisplatin by regorafenib was associated with EGFR and NF-κB inactivation.
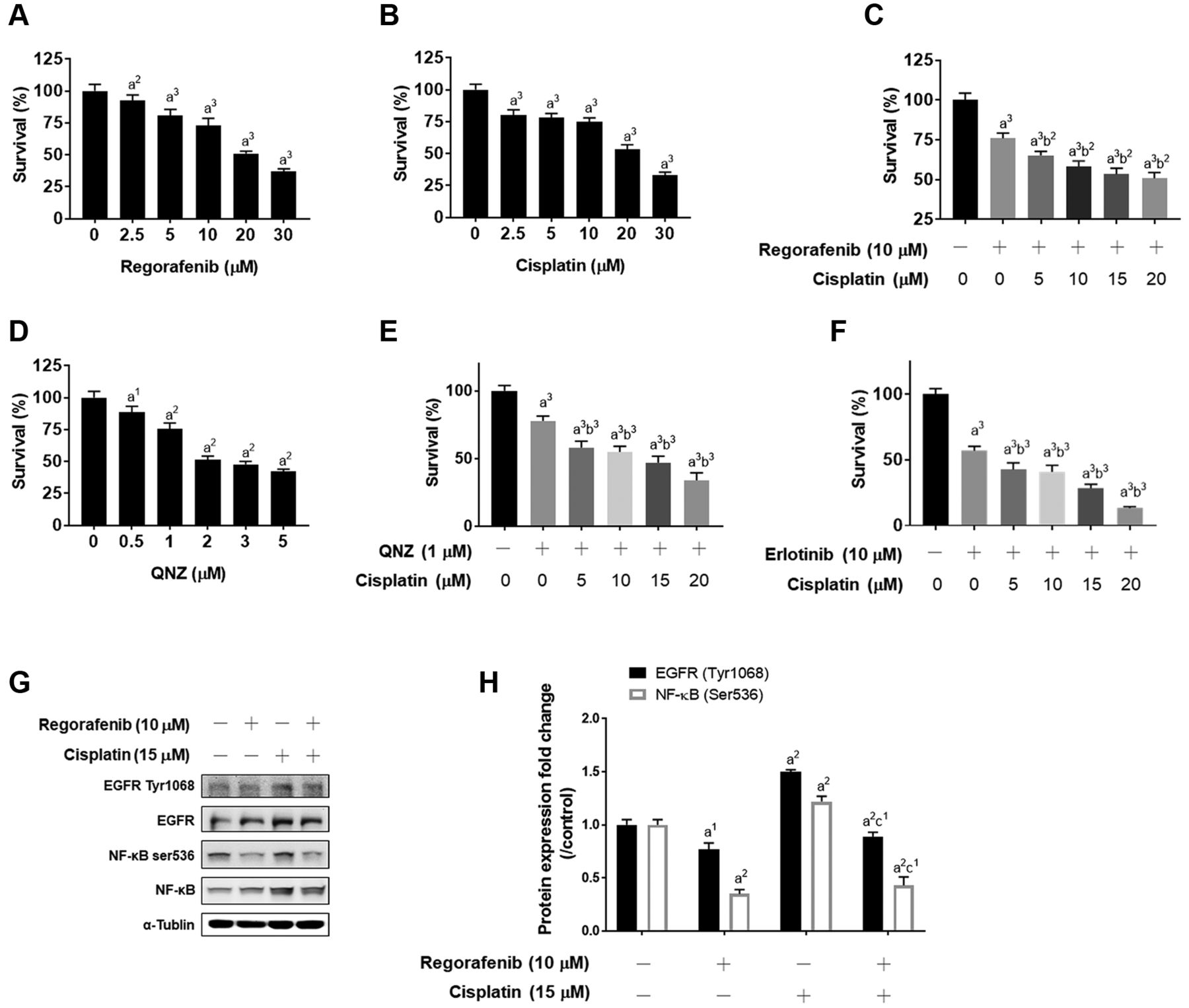
Induction of cytoxicity and inactivation of EGFR/NF-κB was found in regorafenib combination with cisplatin in A549 cells. A549 cells were treated with (A) 0-30 μM regorafenib, (B) 0-25 μM cisplatin, (C) 10 μM regorafenib combined with 0-25 μM cisplatin, (D) 0-5 μM QNZ, (E) 1 μM QNZ combined with 0-25 μM cisplatin and (F) 10 μM erlotinib combined with 0-25 μM cisplatin for 48 h and assayed by MTT. (G-H) The expression pattern and quantification results of EGFR and NF-κB phosphorylation are shown. a1p<0.05, a2p<0.01, a3p<0.005 vs. 0 μM of treatment; b2p<0.01, b3p<0.005 vs. 10 μM regorafenib, 1 μM QNZ or 10 μM erlotinib; c1p<0.05 vs. 15 μM cisplatin).
Regorafenib intensified cisplatin-inhibited invasion and migration of NSCLC cells. To investigate whether regorafenib combined with cisplatin may further inhibit the metastatic potential of A549 cells, we performed invasion and migration transwell assays. As illustrated in Figure 2A, the percentage of migration and invasion capacity of A549 cells was decreased in the combination treatment group as compared to single-treatment group (Figure 3A). In Figure 2B and C, the lowest migration and invasion areas were found in regorafenib combined with cisplatin group. The migration capacity of A549 cells was suppressed by cisplatin combined with regorafenib group as compared to each of the single treatment groups. Also, the number of invading cells was further reduced by the combined treatment compared to the single treatment groups (Figure 3A and C). Furthermore, we investigated whether metastasis-related proteins, such as VEGF, MMP-2, and MMP-9 may be suppressed by regorafenib combined with cisplatin. A significant reduction of VEGF, MMP-2, and MMP-9 was found in the combination treatment group compared to regorafenib and cisplatin single treatment (Figure 3D). In conclusion, regorafenib can facilitate cisplatin-induced metastatic inhibition.
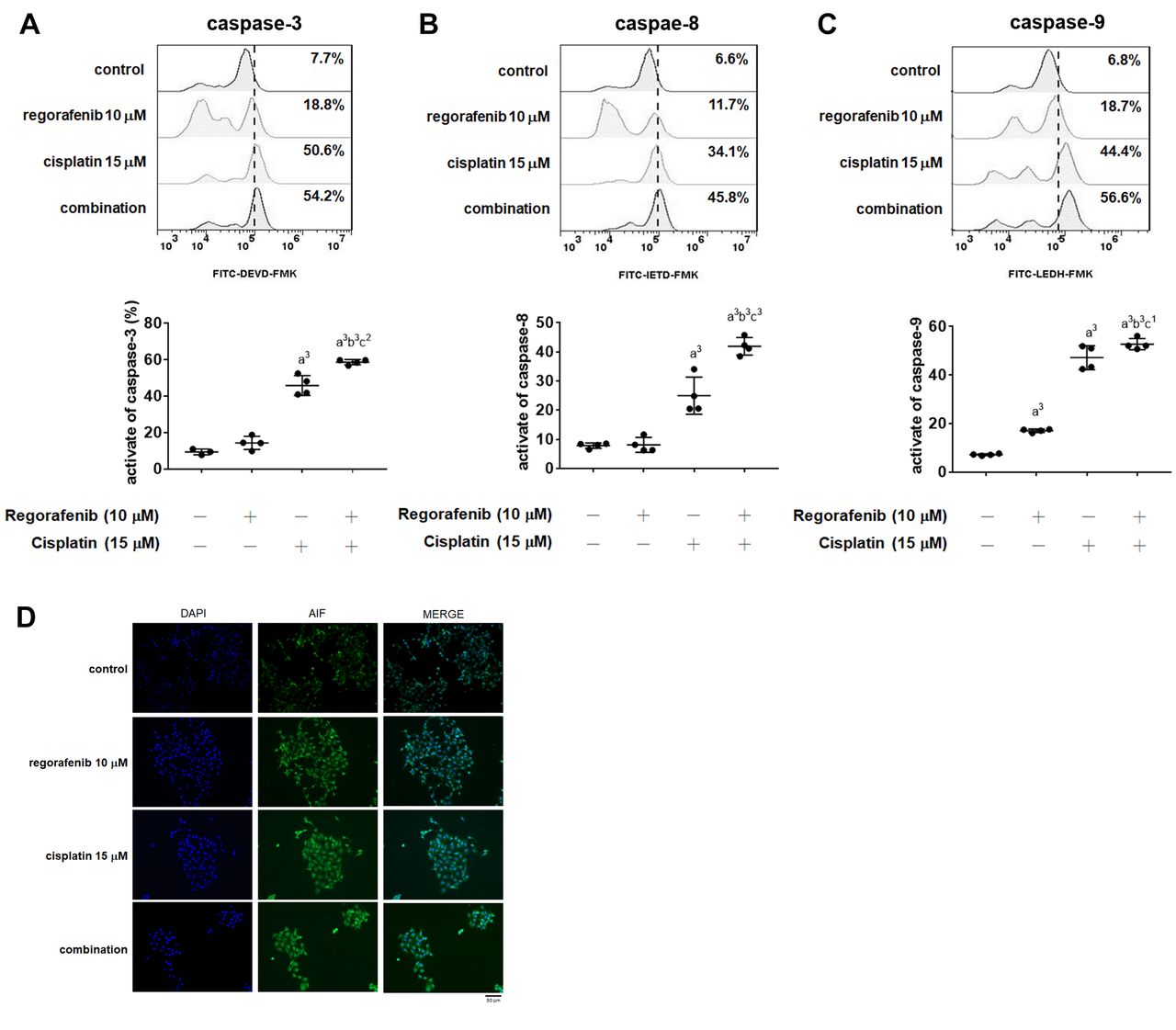
Synergistic activation of death receptor of apoptosis and mithochondria-dependent apoptosis were found in regorafenib combined with cisplatin in A549 cells. A549 cells were treated with 10 μM regorafenib, 15 μM cisplatin and their combination for 48 h. The activation patterns and quantification results of (A) caspase-3, (B) caspase-8, and (C) caspase-9 were assayed by flow cytometry. (D) AIF nuclear translocation pattern of each group is displayed. a2p<0.01, a3p<0.005 vs. 0 μM of treatment; b3p<0.005 vs. 10 μM regorafenib; c1p<0.05, c2p<0.01, c3p<0.005 vs. 15 μM cisplatin scale bar = 50 μM.

{kind=link}
{kind=link}
{kind=link}
Suppression of metastasis effect was found in regorafenib combined with cisplatin in A549 cells. A549 cells were treated with 10 μM regorafenib, 15 μM cisplatin and their combination for 48 h. (A) Migration and invasion pattern of A549 after different treatments are displayed. The quantification of (B) migration area and (C) invasion area after different treatments are presented. (D) The metastasis related proteins expression pattern and quantification bar chart are displayed. a2p<0.01, a3p<0.005 vs. 0 μM of treatment; b2p<0.01, b3p<0.005 vs. 10 μM regorafenib; c2p<0.01 vs. 15 μM cisplatin scale bar = 100 μM.
Regorafenib enhanced cisplatin-triggered intrinsic and extrinsic apoptosis effects in NSCLC cells. To investigate whether regorafenib affects the chemosensitivity and enhances the apoptosis effect of cisplatin, we investigated the activation of apoptosis indicators, such as caspase-3, -8 and -9. As illustrated in Figure 2A-C, the activate forms of caspase-3, -8 and -9 were significantly induced by combining regorafenib and cisplatin for 48 h. These results showing that regorafenib enhances both intrinsic (caspase-8) and extrinsic (caspase-9) apoptosis pathways induced by cisplatin compared to cisplatin alone. After validating caspase-dependent apoptosis, caspase-independent marker, AIF was also investigated by immunofluorescence staining. As shown in Figure 2D, the nuclear translocation of AIF was increased by regorafenib combined with cisplatin. Taken together, regorafenib effectively triggered cisplatin-induced intrinsic and extrinsic apoptosis pathways.
Discussion
Binding of EGFR to its ligands induces rapid tumor progression through initiation of downstream pathways such as RAF/mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) kinase/ERK and phosphoinositide 3-kinases (PI3K)/AKT (22, 23). NF-κB activity is triggered with EGFR signaling and is required for EGFR-driven NSCLC progression (24). The NF-κB signaling cascade triggers expression of downstream effector proteins such as MMP-9, -2, and VEGF leading to lung cancer progression (25). Activation of both EGFR and NF-κB was triggered by cisplatin and associated with chemoresistance in cancers (26).
Geftinib, the first generation of EGFR-TKI, has been indicated to enhance an inhibitory effect of cisplatin through disruption of the EGFR signaling cascade in wild-type EGFR NSCLC cells (6). Our data showed that regorafenib, erlotinib, and QNZ effectively augmented cytotoxicity of cisplatin in A549 cells (Figure 1C, E and F). Notably, cisplatin-increased protein levels of both EGFR Tyr1068 and NF-κB ser536 were significantly inhibited by regorafenib treatment (Figure 1G-H). Based on these results, we suggest that suppression of EGFR/NF-κB signaling contributes to regorafenib-augmented cytotoxicity of cisplatin in A549 cells.
Invasion-associated proteins MMP-2 (72 kDa gelatinase A) and MMP-9 (92 kDa gelatinase B) promote tumor invasion and metastasis through disruption of extracellular matrix (ECM) and induction of epithelial-mesenchymal transition (EMT) (27, 28). High expressions of both MMP-2 and MMP-9 as unfavorable prognostic predictors were correlated with lymph node metastasis and poor prognosis in NSCLC patients (29, 30). Our results demonstrated that the combination of regorafenib and cisplatin not only significantly reduced the number of migration and invasion cells, but also effectively abolished protein levels of both MMP-2 and MMP-9 compared to regorafenib or cisplatin monotherapy (Figure 3). Decreased protein levels of MMP-2 and MMP-9 were condutive to regorafenib and effectively triggered anti-invasion ability of cisplatin in NSCLC cells.
Cisplatin induces tumor cell death through extrinsic and intrinsic apoptotic pathways. Extrinsic caspase-8 and intrinsic caspase-9 pathways activate caspase-3 and promote release of AIF from mitochondria resulting in formation of apoptotic DNA fragmentation (31-33). Decreased expression and activation of caspase-8, -9, and AIF have been found to be related to cisplatin insensitivity in NSCLC cells (34-36). Our results showed that regorafenib effectively enhanced cisplatin-induced caspase-3, -8, and -9 activation (Figure 2A-C). Furthermore, cisplatin-induced AIF nuclear translocation was increased by regorafenib treatment (Figure 2D). Based on these results we suggest that regorafenib enhanced cisplatin-triggered apoptosis via caspase-dependent and - independent pathways in NSCLC cells.
In conclusion, this study showed that regorafenib enhanced cisplatin-induced tumor cell growth inhibition and showed ability in invasion suppression in A549 cells. Regorafenib effectively enhanced cisplatin-inhibited cell growth and invasion in A549 cells. In addition, regorafenib also promoted cisplatin-induced apoptosis through caspase-dependent and -independent pathways. We suggest that induction of apoptosis and inhibition of EGFR/NF-κB signaling was associated with the anti-NSCLC effect of regorafenib for sensitizing NSCLC cells to cisplatin.
Acknowledgements
This study was supported by Taichung Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Taichung, (ID: TTCRD109-16) and Chang Bing Show Chwan Memorial Hospital, Changhua (ID: BRD-110013), respectively. Experiments and data analysis were performed in part through the use of the Medical Research Core Facilities Center, Office of Research & Development at China Medical University, Taichung, Taiwan, R.O.C.
Footnotes
This article is freely accessible online.
* These Authors contributed equally to this study.
Authors’ Contributions
JYW, YSW, and FTH performed the experiments. JYW, YSW, YCC prepared the initial version of the article. JYW, YSW, YCC, FTH, and ITC designed the study, performed the literature review, and prepared the final versions of the article.
Conflicts of Interest
The Authors declare no competing financial interests regarding this study.
- Received May 19, 2021.
- Revision received June 4, 2021.
- Accepted June 15, 2021.
- Copyright © 2021 International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved