

Abstract
Background/Aim: Casein kinase 2 (CK2) which sustains multiple pro-survival functions in cellular DNA-damage response, is strictly regulated in normal cells but elevated in cancer. CK2 is considered as a potential therapeutic target, and its inhibition has been associated with radiosensitization in mammalian cells in vitro. Here, we investigated potential radiosensitization by CK2 inhibition in vivo. Materials and Methods: The effect of CK2 inhibition in vivo was investigated in human WiDr-xenograft tumours grown subcutaneously on BALB/c nu/nu mice with and without fractionated irradiation. CK2 inhibition was performed using the specific inhibitor tetra-bromobenzotriazole (TBB). Histological examinations included staining for apoptosis and double-strand breaks. Results: Both TBB treatment alone and radiation alone significantly reduced tumour growth, which was reflected by increased apoptosis rates. However, TBB treatment did not boost radiation-induced tumour growth suppression in combined treatment, although the apoptosis rate increased and repair of double-strand breaks was reduced. This was in stark contrast to previous data on in vitro radiosensitization. Conclusion: The absence of radiosensitization by CK2 inhibition should be investigated in different tumour models.
- CK2 inhibition
- tetra-bromobenzotriazole
- TBB
- irradiation
- human WiDr tumours
Protein kinase CK2 occurs ubiquitously. It is a highly conserved serine/threonine kinase with a broad spectrum of target proteins that are up-regulated regularly during embryogenesis of vertebrates. Most of these proteins play an important role in signal transduction and gene expression for factors with pro-survival functions (1-3). CK2 most often occurs as a heterotrimeric holoenzyme. In mammalian cells, its basal activity is driven by intramolecular interactions in one of two catalytic isoforms: CK2α or CK2α´. Bonding to a dimer of the regulatory subunit CK2β (ααβ2, α´α´β2, or αα´β2) regulates the process (1, 4). Most malignant tumours display CK2 dysregulation, with especially high levels found in the nuclear compartment (1, 5, 6), but the exact role of CK2 in tumorigenesis is yet to be fully determined. There is, however, some evidence suggesting that CK2 suppresses apoptosis via tumour-suppressor regulation and oncogene activity (6). CK2 is, therefore, a legitimate target for tumour-specific therapies (7) when treating diseases, such as malignant gliomas (8), acute lymphoblastic cells (9), prostate carcinoma (10), or breast cancer (11).
Limited data are available on the phenotypic expression of these CK2-dependent interactions. Some authors have shown that radiation sensitivity increases when a mutation of the CK2-binding site is present on the X-ray repair cross-complementing protein 4 (XRCC4). This applies to both clonogenic survival and de-phosphorylation of the H2A-histone family member X (γ-H2AX) (12). However, when examining the immediate repair of DNA fragments, our team was unable to detect any reduction in kinetics of repair of double-stranded breaks (DSBs) in DNA caused by CK2 inhibition in human colon carcinoma cells (WiDr) using the chemical inhibitor 4,5,6,7-tetra-bromobenzotriazole (TBB) (13, 14). However, we did detect reduced de-phosphorylation of γ-H2AX due to TBB-mediated CK2 inhibition. We also found that CK2 inhibition led to low but significant clonogenic radiosensitivity, which may in part be caused by a reduction of the DNA-damage response due to CK2 inhibition. These CK2-dependent radiosensitizing effects were tumour-specific when a dose of up to 3 Gy was applied (14).
The aim of this investigation was to investigate radiosensitization induction by CK2 inhibition in WiDr colorectal adenocarcinoma cells in an in vivo model, which has previously been confirmed in vitro, as well as to explore the effect of CK2 inhibition on γ-H2AX phosphorylation and apoptosis induction in tumour tissue.
Materials and Methods
Cell lines and culture conditions. The origin and culture conditions of the human WiDr cells (colon adenocarcinoma) have been described elsewhere (15). WiDr cells (Tumorbank of the German Cancer Research Center, Heidelberg, Germany) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal calf serum (Biochrom, Berlin, Germany) and 1% L-glutamine (Serva, Heidelberg, Germany). Cells were grown as monolayers in 6% humidified CO2/air at 37°C. Under these conditions, tumour cell cultures exhibited doubling times of 28 h, with plating efficiencies ranging from 70-90%.
Drug treatment and irradiation. Aliquots of 10 mM stock solution of the CK2 inhibitor, TBB (Calbiochem, Merck, Darmstadt, Germany), in dimethylsulfoxide (DMSO) were stored at –20°C. Further dilutions for in vivo experiments were prepared with phosphate-buffered saline (PBS) (1×). Mock treatments were performed by adjusting the respective DMSO/PBS volumes to those of the TBB samples. In vivo irradiation was conducted using a linear accelerator (Siemens, Munich, Germany) with 6 MV photons at a dose rate of 2.5 Gy/min.
Xenograft tumour model. Animal studies were performed according to the rules for care and use of experimental animals and were approved by the local and governmental Animal Care Committees (Regierungspräsidium Karlsruhe, Baden-Württemberg, Germany; approval numbers: AZ 35-9185.81/G-8509 and KZ 8911220007311). WiDr xenografts were established by injecting 5×106 WiDr cells subcutaneously into the right hind limb of 6- to 8-week-old BALB/c athymic nude mice, each weighing 20 g (Charles River Laboratories, Sulzfeld, Germany). Tumour treatments (radiation and TBB) were started when the tumour volume reached approximately 100 mm3 (day 0), as measured with callipers and calculated using the following formula:
Volume = length × width2 × 0.5
The animals were then randomised at 16 mice per group (with three mice per group scheduled for histology on day 4 after treatment commenced) into the following four groups: DMSO control, TBB only, DMSO control plus irradiation, and TBB plus irradiation. TBB diluted in DMSO was administered intraperitoneally in PBS (150 mg/kg or 1 mM) twice daily for 5 consecutive days (day 0-4). Tumours were irradiated based on a fractionated schedule (5×2 Gy) beginning on day 0 for 5 consecutive days (days 0-4), while all other parts of the mice were spared (Figure 1).

Schema of the chronological sequence of the in vivo WiDr xenograft model investigation. TBB: 4,5,6,7-Tetra-bromobenzotriazole.
Immunohistology. For immunohistology, animals were sacrificed on day 4 (the last day of treatment), 6 h after radiotherapy. Apoptosis and γ-H2Ax phosphorylation were detected via indirect immunofluorescent examination of frozen tumour sections, performed according to standard protocols.
Apoptosis: Cryostat tumour sections were fixed in 4% paraformaldehyde in PBS for 1 h at room temperature. Terminal desoxynucleotidyl transferase-mediated deoxy-uracil triphosphate biotin nick-end labelling (TUNEL) assays were performed using an in situ Cell Death Detection Kit (Roche Diagnostics GmbH, Mannheim, Germany) according to the manufacturer’s instructions. The number of TUNEL-positive cells per field of view (average cell count of 510; 40× magnification) was used to quantitatively compare treatment effects. The cell density per region of interest was determined by counting 4’,6-diamidine-2-phenylindole-stained nuclei. Digital fluorescent images were obtained using a Nikon Eclipse E600 microscope (Nikon, Dusseldorf, Germany) equipped with a Nikon digital sight DS-U1 camera.
γ-H2AX: Cryostat tumour sections were fixed in 4% paraformaldehyde in PBS for 1 h at room temperature and permeabilised [20 mM HEPES (pH 7.4), 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose, and 0.5% Triton X-100 for 5 min at 4°C; Sigma-Aldrich, Taufkirchen, Germany)] Antibodies were mouse anti-γH2AXSer139 (Merck Millipore, Burlington, MA, USA) and Alexa Fluor 680 goat anti-mouse secondary antibodies (Invitrogen, Thermo Fisher, Waltham, MA, USA). Primary antibodies were incubated with anti-γH2AX for 40 min at 37°C, and a 1:500 dilution (2 μg/ml) in PBS supplemented with 2% bovine serum fraction V albumin (Sigma-Aldrich) was performed. Incubations with fluorochrome-labelled secondary anti-mouse antibodies were performed at 37°C for 20 min, and 1:200 dilution (10 μg/ml) in 2% bovine serum fraction V albumin was performed. Nuclei were counterstained in Vectashield mounting medium with 4’,6-diamidino-2-phenylindole (Vector Laboratories, Peterborough, UK) for 5 min at 20°C. The number of γH2AX-positive cells per field of view (average cell count of 1,014; 20× magnification) was used to quantitatively compare treatment effects. Digital fluorescent images were obtained using a Nikon Eclipse E600 microscope equipped with a Nikon digital sight DS-U1 camera (Nikon).
Statistical analysis. Differences between experimental groups were analysed for statistical significance using Student’s t-test. A p-value of less than 0.05 was considered significant.
Results
Tumour growth delay. Single TBB treatments significantly reduced tumour growth compared to DMSO controls (please see Figure 2). Compared to the DMSO control group, the only TBB-treated group showed a mean tumour growth delay of 1.7 days (10.2 vs. 11.9 days) and 3.5 days (20.8 vs. 24.3 days) in the time taken to attain 4- and 8-fold tumour volumes, respectively, after the commencement of treatment.

In vivo growth of subcutaneous WiDr human colon carcinoma xenografts in BALB/c nu/nu mice. Each group included 16 mice at the beginning of treatment: Dimethyl sulfoxide (DMSO) control, 4,5,6,7-tetra-bromobenzotriazole only (TBB), DMSO control plus irradiation (RT), and TBB plus RT. TBB treatment (150 mg/kg, 1 mM intraperitoneal) was performed twice daily (days 0-4), and fractionated irradiation (2 Gy single fraction, 10 Gy cumulative) was applied to the tumour daily (days 0-4). Date are the mean relative tumour volume normalised to that on the day that therapy began (day 0). Bars indicate standard errors. *Statistically significantly different at p<0.05.
As expected, irradiation treatment caused marked and significantly delayed tumour growth compared to the DMSO control group (Figure 2). Interestingly, in the combination-treated group, TBB-mediated CK2 inhibition did not further enhance radiation-induced tumour growth delay (Figure 2).
Tumour immunohistology. After 5 days of treatment with TBB twice a day and fractionated radiotherapy (5×2 Gy), the mean number of γH2AX-positive tumour cells per field of view increased significantly among irradiated WiDr tumours from mice simultaneously treated with TBB (Figure 2; p<0.05). TBB treatment alone did not significantly influence the number of γH2AX-positive WiDr tumour cells in vivo. As anticipated, the group treated with irradiation alone exhibited a significantly increased number of γH2AX-positive tumour cells compared to non-irradiated controls (Figures 3 and 4).

Immunohistochemistry for in vivo signalling of the H2A-histone family member X (γ-H2AX) in WiDr human colon carcinoma xenograft in BALB/c nu/nu mice after different treatments. A: Dimethyl sulfoxide (DMSO) control. B: 4,5,6,7-tetra-bromobenzotriazole only (TBB) only. C: DMSO control plus irradiation. D: TBB plus irradiation.

Immunohistochemical evaluation of the effect of each treatment on γ-H2AX signalling in WiDr human colon carcinoma xenograft in BALB/c nu/nu mice after different treatments: Dimethyl sulfoxide (DMSO) control; 4,5,6,7-tetra-bromobenzotriazole only (TBB) only; DMSO control plus irradiation (RT); TBB plus RT. Quantitative comparison of γ-H2AX-positive cells per field of view for excised tumour sections from the four groups of animals. A field of view (20×) showed, on average, 1,014 cells. Data represent mean values and standard deviations from at least four fields of views from at least three different mice. *Statistically significantly different at p<0.05.
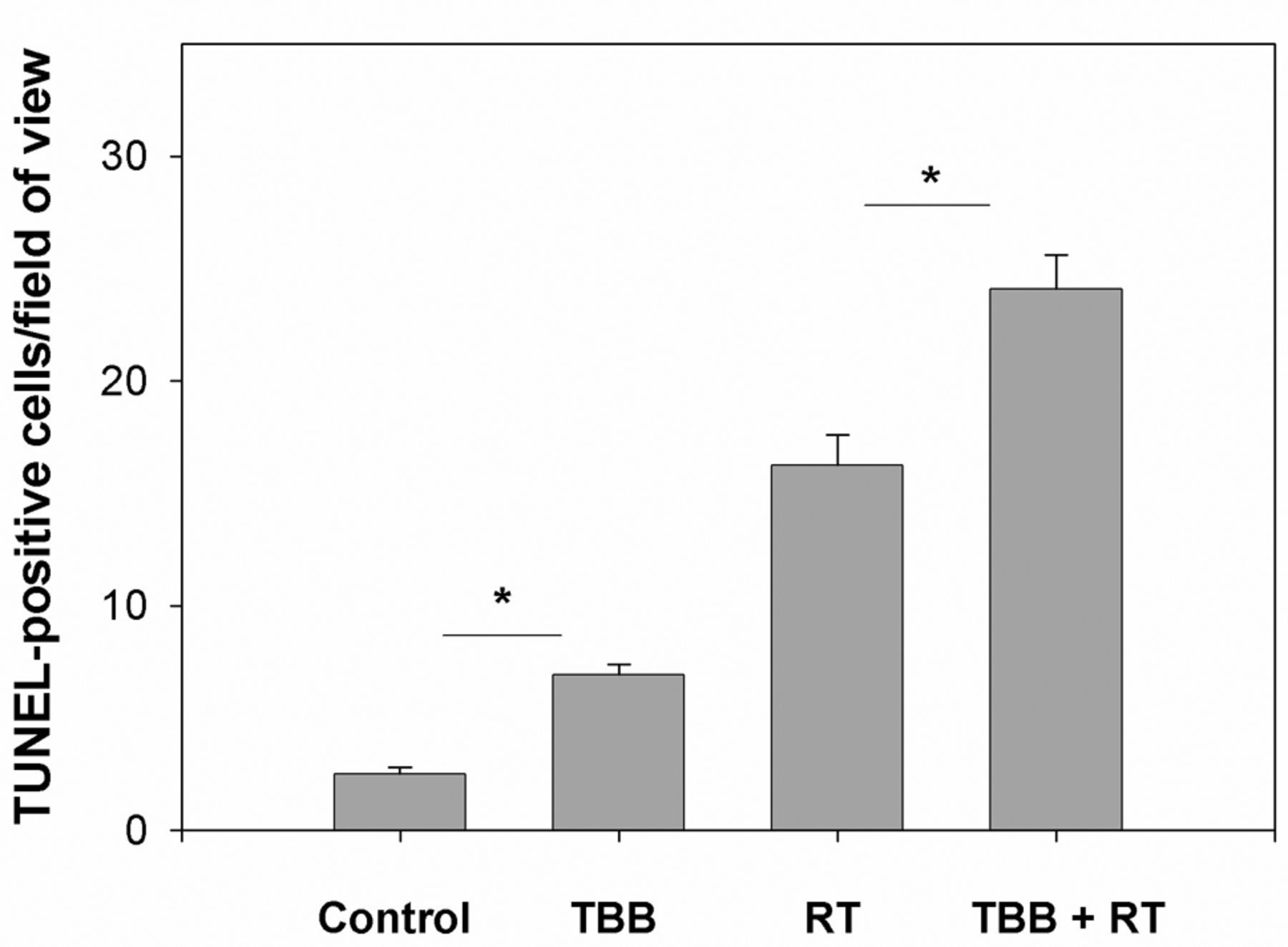
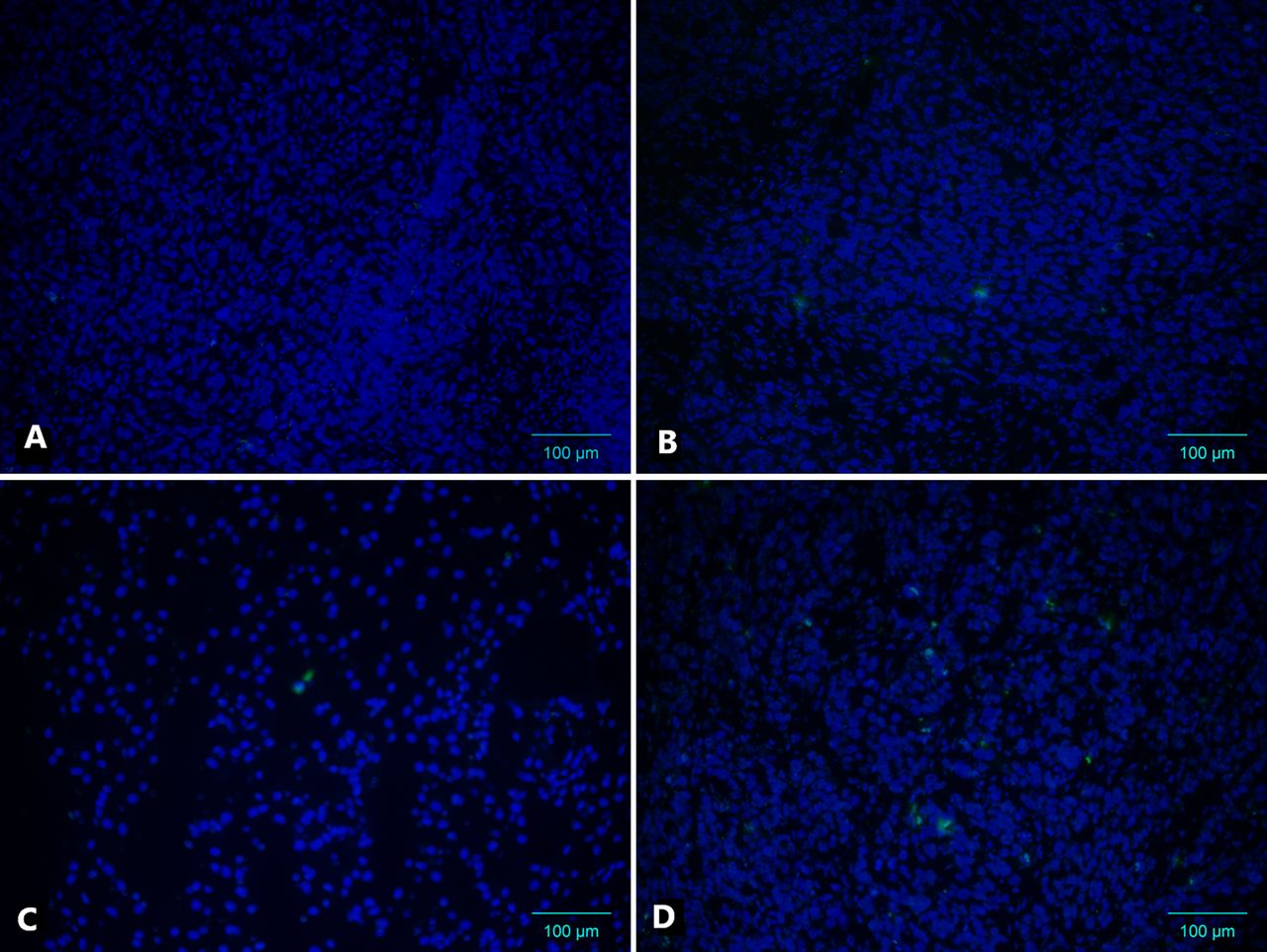
In vivo apoptosis rates were also measured on day 4 (end of irradiation; cumulative dose of 10 Gy). The group treated with TBB alone showed a significant increase in the mean number of TUNEL-positive tumour cells compared with the untreated DMSO control group (Figures 5 and 6; p<0.05). As expected, the group treated with irradiation alone exhibited a significant increase in apoptosis compared with the control and TBB-only treatment groups. Additionally, TBB treatment accounted for a significant increase in the mean number of apoptotic cells in irradiated WiDr tumours (Figures 5 and 6; p<0.05).

Terminal desoxynucleotidyl transferase-mediated deoxy-uracil triphosphate biotin nick-end labelling assay of WiDr human colon carcinoma xenografts in BALB/c nu/nu mice after different treatments. A: Dimethyl sulfoxide (DMSO) control. B: 4,5,6,7-tetra-bromobenzotriazole only (TBB) only. C: DMSO control plus irradiation (RT). D: TBB plus RT.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Quantitative comparison of terminal desoxynucleotidyl transferase-mediated deoxy-uracil triphosphate biotin nick-end labelling (TUNEL)-positive cells per field of view for sections of excised tumour from WiDr human colon carcinoma xenograft in BALB/c nu/nu mice after different treatments: Dimethyl sulfoxide (DMSO) control; 4,5,6,7-tetra-bromobenzotriazole (TBB) only; DMSO control plus irradiation (RT); TBB plus RT. A field of view (40×) showed, on average, 510 cells. Data represent mean values and standard deviations from at least four fields of view from at least three different mice. *Statistically significantly different at p<0.05.
Discussion
In many previous in vitro studies, tumour cells displayed growth inhibition mediated by CK2 inhibition. Studies have also shown reduced clonogenic survival following radiation in vitro and enhanced antitumor effects in vivo in some tumour models, suggesting a strong and promising radiosensitization potential for CK2 inhibiting agents (7-9, 14).
Our in vivo experiments in a model of tumour (WiDr) growing subcutaneously in mice showed that CK2 inhibition reduced tumour growth. As expected, radiation alone also caused marked tumor growth delay. Surprisingly however, we found here that TBB-mediated CK2 inhibition did not enhance radiation effects and did not enhance radiation-induced tumor growth delay. This was unexpected, since in previous in vitro experiments, we had found a strong radiosensitizing effect in vitro e.g. in clonogenic survival assays in TBB-treated WiDr cells (14). Likewise, in an in vivo BALB/c nude mouse model, Li et al. reported a significant reduction in growth of irradiated non-small-cell lung cancer (H460) following CK2 inhibition by specific inhibitor quinalizarin (16). These different results might be attributed to the use of different CK2 inhibitors, i.e. TBB versus quinalizarin, with the well-documented specific inhibitory effects of quinalizarin on CK2 (14, 16). Furthermore, Li et al. injected quinalizarin diluted in DMSO intraperitoneally (35 mg/kg/day) for 3 consecutive days (days 9-11) and twice a week after single-shot irradiation (10 Gy) on day 12. Thus, quinalizarin was not administered simultaneously with radiation but sequentially, suggesting that the time interval produced an additive effect. In our in vivo experiment, the CK2 inhibitor, TBB (150 mg/kg twice daily for 5 consecutive days) was administered simultaneously with radiotherapy (2 Gy daily and 10 Gy of cumulative irradiation), which then would mean that the immediate combination effect is not beneficial, in fact it suggests that TBB somehow reduces the radiation effect on the tumor or vice versa.
The specificity of the chemical kinase inhibitors for certain target enzymes is most likely particularly important here. In previous in vitro testing, TBB showed a strong selectivity for CK2 among ~80 kinases. In total, only three other kinase groups were inhibited by TBB with a comparable level of efficiency (13). These three do not participate in DSB repair; although two of them (HIPK2 and DYRK2) regulate the p53-dependent differential transactivation of growth-inhibiting genes versus pro-apoptotic genes, depending on the degree of DNA damage (17). The half-maximal inhibitory concentration (IC50) determined in vitro for TBB was 0.15 μM (13), a concentration that is 100 times lower than those obtained in tests where CK2 protein extracts were obtained from cells pre-treated with TBB and used to phosphorylate synthetic CK2 target peptides in vitro (18, 19).
Immunocytochemistry using CK2α´ antibodies showed that CK2 was expressed in WiDr cells and exhibited a strong tendency to collect in the perinuclear compartment of the tumour cells (14). The inhibitory effect of TBB on CK2 in WiDr cells was determined and tested using a functional in vitro assay. In this test, the phosphorylation status of XRCC1 protein was analysed 12 h after adding TBB (20 μM). A distinct reduction in XRCC1 phosphorylation due to TBB was evident in the fluorescence-activated cell sorting histogram analysis (14). In our in vivo experiment, TBB was administered intraperitoneally (150 mg/kg or 1 mM) twice daily for 5 consecutive days and could be considered effective because TBB monotherapy showed efficacy regarding tumour growth delay.
The discrepancy in our results compared with others may be due to the different tumour models. We implanted WiDr adenocarcinoma cells from human colorectal tumours into BALB/c nude mice, whereas Li et al. implanted tumour cells from human non-small-cell carcinoma (16) implying different interactions with the surrounding microenvironment (20).
We also found that TBB delayed the reduction in radiation-induced H2AX foci in WiDr cells following radiation. These findings are concordant with previous reports from in vitro studies; CK2 inhibition modulated the cellular response following irradiation, but it did not reduce the physical DSB-repair rate, as measured by pulsed-field gel electrophoresis (14, 21-22).
γ-H2AX foci are broken down by in situ de-phosphorylation or histone exchange followed by de-phosphorylation of the stored γ-H2AX histone. Phosphatase PP2A seems to play a key role in dephosphorylating γ-H2AX in mammalian cells (23) and has been shown to interact directly with the α-kinase catalytic subunit of CK2, which becomes phosphorylated and activated (24). This suggests that CK2 may play a role in γ-H2AX turnover. Overall, our results provide evidence that TBB-mediated CK2 inhibition in vivo leads to delayed γ-H2AX signal involution.
Regarding the established pro-survival effect of CK2, the impact of CK2 inhibition on the apoptosis rate, with and without irradiation, is a subject of interest. Previous data suggest that CK2 inhibition often leads to increased apoptosis rates in tumour cells, in vitro and in vivo (16, 25-27), and can significantly boost radiation-induced induction of apoptosis (16, 28). In our in vivo experiment, TBB-mediated CK2 inhibition also significantly increased the apoptosis rate in WiDr tumours compared to the untreated control group, as well as a significant boost in the radiation-induced apoptosis rate. We found that the increase in apoptosis caused by TBB-mediated CK2 inhibition, when compared to the untreated control group, correlated with significant retardation of tumour growth. However, further CK2 inhibition in the irradiated group did not enhance tumour growth retardation.
Conclusion
In the in vivo tumour model used here, CK2 inhibition led to significant tumour growth retardation compared to the untreated control group. However, we did not observe tumour-specific in vivo radiosensitization in human colorectal adenocarcinoma (WiDr) due to TBB-mediated CK2 inhibition. Our results were contrary to previous studies, which used a different tumour model, indicating tumour type-specific interaction. Moreover, our data also caution against the direct translation of in vitro tumour radiosensitization effects to the in vivo situation, which might be different.
Acknowledgements
The Authors wish to thank Alexandra Tietz Dalfuß, Marina Szymbara, and Sylvia Trinh for their excellent technical assistance.
Footnotes
Authors’ Contributions
F.Z., P.H., K.W., and J.D. initiated and supervised the project. F.Z. collected data. F.Z., and P.H. carried out the data analysis. F.Z., H.H., P.H., and J.D. interpreted the experimental data and were responsible for creating figures. F.Z. and P.H. wrote the article with input from all Authors. All Authors were involved in article revision.
This article is freely accessible online.
Conflicts of Interest
The Authors declare that there are no conflicts of interest in regard to this study.
- Received October 3, 2020.
- Revision received October 17, 2020.
- Accepted October 21, 2020.
- Copyright© 2021, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved