

Abstract
Background/Aim: Tolerability and safety of trastuzumab emtansine (T-DM1) was investigated in Japanese patients with HER2-positive advanced breast cancer who were previously treated with chemotherapy and trastuzumab. Patients and Methods: Patients with inoperable or recurrent breast cancer who were previously treated with chemotherapy and trastuzumab in adjuvant and/or metastatic disease were included. T-DM1 3.6 mg/kg was administered intravenously every 21 days. The administration dosage or schedule of T-DM1 was modified based on laboratory tests on the administration day. Results: Among 232 patients analyzed, adverse events were reported in 228 patients (98.3%); five patients (2.2%) discontinued due to adverse events and twenty patients (8.6%) had serious adverse events. The most commonly reported grade ≥3 adverse event of special interest was thrombocytopenia (69 patients; 29.7%), followed by hepatotoxicity (26 patients; 11.2%). Conclusion: T-DM1 was well tolerated in Japanese patients with HER2-positive advanced breast cancer and no new safety signals were observed.
- Breast cancer
- HER2
- trastuzumab emtansine (T-DM1)
- anti-HER2 therapy
- Japanese patients
The human epidermal growth factor receptor 2 (HER2) has been shown to play an important role in the development and progression of certain types of breast cancer. HER2 is overexpressed in ~20% of patients with breast cancer and is associated with a more aggressive tumor growth and poor clinical outcome (1-3). Since trastuzumab, an anti-HER2 humanized monoclonal antibody, demonstrated the significant survival benefit in patients with HER2-positive metastatic breast cancer (4), combination of anti-HER2 therapy with standard chemotherapy has been used as an effective therapeutic approach for patients with HER2-positive advanced breast cancer. In addition, dual blockade of HER2 with combination of pertuzumab and trastuzumab plus chemotherapy showed significant improvement in overall survival compared to trastuzumab plus chemotherapy (5). To date, several novel anti-HER2 therapeutic agents, including trastuzumab, lapatinib, pertuzumab, and trastuzumab emtansine, have been developed.
Trastuzumab emtansine, known as T-DM1, is an antibody drug conjugate incorporating trastuzumab with microtubule-inhibitory agent DM1 (derivative of maytansine); the anti-HER2 antibody and cytotoxic agent are conjugated via a stable linker (6, 7). T-DM1 is designed to deliver the cytotoxic agent specifically to HER2-positive cells, that may improve the clinical benefits of anticancer therapy of trastuzumab with DM1 and minimize the exposure to normal tissues. Phase III study (EMILIA) demonstrated that T-DM1 significantly prolonged progression-free and overall survival with a more favorable safety profile than lapatinib plus capecitabine in patients with HER2-positive advanced breast cancer who were previously treated with trastuzumab and taxane (8). Another phase III study (TH3RESA) demonstrated that T-DM1 prolonged progression-free and showed survival benefit with less high-grade toxicity compared to a treatment of physician's choice in patients with HER2-positive advanced breast cancer who were previously treated with both trastuzumab and lapatinib in the advanced setting and a taxane in any setting, and had disease progression after at least two regimens of HER2-directed therapy (9).
In Japan, T-DM1 was approved with the recommended dose of 3.6 mg/kg given as an intravenous infusion every 3 weeks according to the results of clinical studies conducted in Japan (10, 11). The most common grade 3/4 adverse events of T-DM1 include thrombocytopenia, and hepatotoxicity (including nodular regenerative hyperplasia, aspartate aminotransferase [AST] increased and/or alanine aminotransferase [ALT] increased) (10). The incidence of grade 3/4 thrombocytopenia (22%) and AST/ALT increased (14%/8%) in Japanese patients (10) tends to be higher than Western population (grade ≥3 thrombocytopenia, 4.7% to 12.9%; grade ≥3 AST increased, <2% to 6.0%, grade ≥3 ALT increased <2% to 2.9%) (8, 9, 12, 13). As further evaluation of safety of T-DM1 in Japanese patients is valuable, we conducted another phase II study (JO29317) to investigate the tolerability and safety, including the incidence of distinct adverse events of T-DM1 in Japanese patients with pre-treated HER2-positive advanced breast cancer under the settings that were similar to routine clinical practice.
Patients and Methods
Patients. Eligible patients had inoperable or recurrent breast cancer and were previously treated with chemotherapy and trastuzumab in adjuvant and/or metastatic disease. Inclusion criteria were age of ≥20 years, and confirmed HER2-positive status by immunohistochemical analysis (with 3+ indicating positive status), fluorescence in situ hybridization (with an amplification ratio ≥2.0 indicating positive status), or both. Patients with Eastern Cooperative Oncology Group performance status of 0-3 were also included. Additional eligibility criteria included adequate bone marrow, hepatic and renal function, as defined by the laboratory tests including platelet count ≥75,000/μl, total bilirubin levels ≤1.5 × the upper limit of normal, and AST/ALT levels ≤5 × the upper limit of normal.
Major exclusion criteria included prior treatment with T-DM1, having experienced a serious hypersensitive reaction to trastuzumab, left ventricular ejection fraction (LVEF) of less than 45% at baseline, a history of serious cardiac disease or dysfunction, central nervous system metastases, and peripheral neuropathy of grade 3 or higher (according to the National Cancer Institute Common Terminology Criteria for Adverse Events [CTCAE] version 4.03, Japanese version).
Study design and measures. The present phase II study is a single arm, multi-center study, conducted between December 2013 and August 2014. The study protocol and all amendments were approved by local Ethic Committees or the institutional review board at each study site. This study was conducted in accordance with the Good Clinical Practice guidelines and the Declaration of Helsinki, as well as other applicable regulatory requirements. All study participants provided written informed consent prior to study entry. The present study has been registered with the Japan Pharmaceutical Information Center (ID: JapicCTI-132395, registered on December 26, 2013).
Patients received a starting dose of 3.6 mg/kg T-DM1 intravenously every 21 days and were followed up until the 28th day after the last dose of T-DM1. Administration of T-DM1 was based on the criteria of dose modification; the first step of dose reduction was to 3.0 mg/kg, and the second step was to 2.4 mg/kg; the next step was discontinuation of T-DM1 therapy. Dose escalation was not allowed after a dose reduction. T-DM1 was administered for at least 3 cycles; therapy was continued until the development of unmanageable toxicity, death, or withdrawal of consent. The study continued until T-DM1 became available for clinical practice at the study site. The administration dosage or schedule of T-DM1 was modified based on platelet count, AST/ALT levels, or total bilirubin levels on the administration day based on the consideration of the abnormal values of platelet count and AST/ALT levels on day 8 of each cycle tended to normalize on the day 1 of the next cycle (14). The administration of T-DM1 was discontinued due to grade 4 AST/ALT increased, grade 2 AST/ALT increased and total bilirubin levels >2 × the upper limit of normal, grade 4 total bilirubin increased, nodular regenerative hyperplasia, or grade ≥3 hypersensitivity/infusion reaction, while it was ceased due to grade 3/4 thrombocytopenia, grade 3 AST/ALT increased, grade 2/3 total bilirubin increased, or grade 3/4 peripheral neuropathy.
The primary outcome of the present study was safety; the incidence of selected adverse events of special interest included thrombocytopenia, hepatotoxicity, peripheral neuropathy, hypersensitivity/infusion reaction, pneumonitis, and cardiac dysfunction. The secondary outcome included the incidence of overall adverse events (any untoward or unfavorable medical occurrence) and serious adverse events (any adverse event that results in death, is life threatening, requires or prolongs hospitalization, causes persistent or significant disability or incapacity), and changes in laboratory tests and vital signs. In addition, subgroup analyses were performed. Modification of administration dosage and schedule of T-DM1 was based on laboratory values on day 8 of cycle 1 in the previous phase II study JO22997 in Japan (10). However, in this study, patients were permitted to receive full dose of T-DM1 on day 1 of cycle 2 according to the laboratory values on that day; tolerability was also evaluated in these patients even though they met criteria for modification of administration dosage and schedule of T-DM1 stated in the JO22997 study. Furthermore, the relationship between the severity of thrombocytopenia and haemorrhage was analyzed.
Information on the baseline of patients characteristics, including age, HER2 and hormone receptor status, hepatic function, haematological information, vital signs, medical history, sites of metastases, and physical condition, were collected at baseline. The LVEF was measured with echocardiography or multiple-gated acquisition scanning before study entry and every 4 cycles thereafter. Information on the laboratory and haematological test values were collected on days 1, 8, and 22 of cycles 1-3, day 1 of cycle 4 and thereafter, and an additional assessment was performed 28 days after the last dose of T-DM1. Vital signs and physical examination findings were also assessed on regular basis. All adverse events were monitored continuously and were coded using the Japanese version of the Medical Dictionary for Regulatory Activities (MedDRA/J) version 16.1 System Organ Class and Preferred Term, and were graded according to the CTCAE version 4.03.
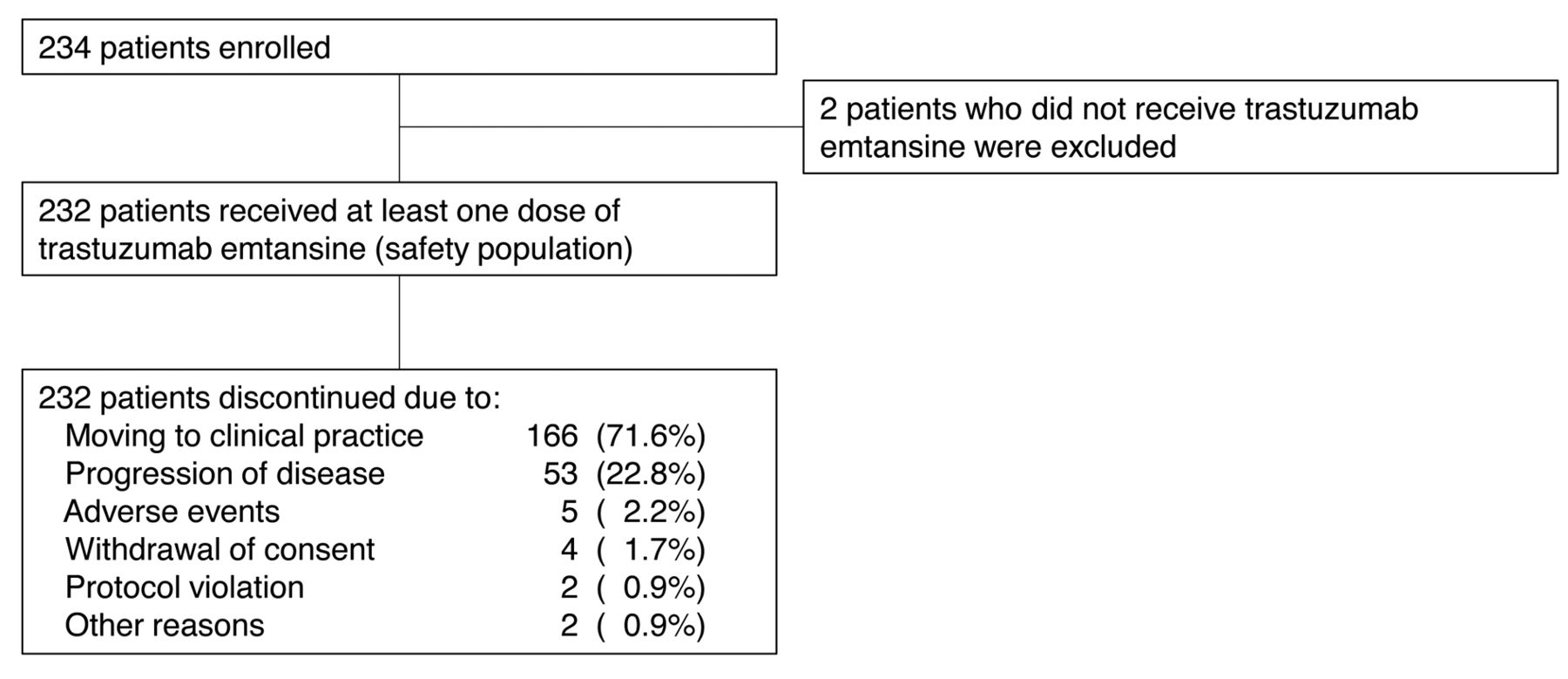
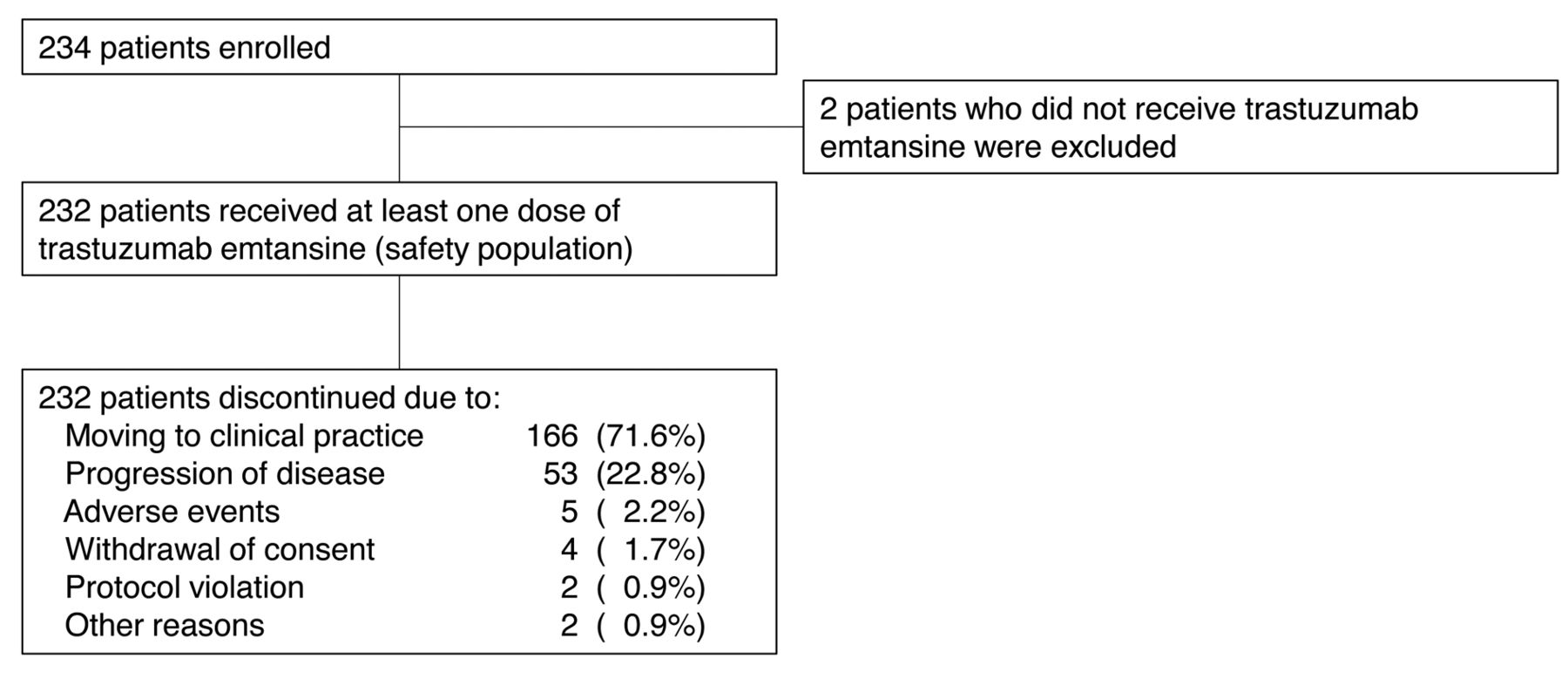
Patient disposition.
Analytical methods. This study was an exploratory study with an enrollment target of approximately 250 patients. The primary analyses of safety included all enrolled patients who received at least one dose of T-DM1. Continuous measurements were summarized by mean or median, and categorical data were summarized by frequency or percentages.
Results
Patients. A total of 234 patients were enrolled at 68 sites in Japan between December 19, 2013 and April 25, 2014, and the last follow-up was performed in August 7, 2014. Among those enrolled in this study, 232 patients received at least one dose of T-DM1 (safety population). During the study period, all 232 patients in the safety population discontinued the study (Figure 1). The majority of them (166 patients; 71.6%) discontinued the study and continued treatment in routine clinical practice after T-DM1 became available on the market in Japan. Among the safety population, the median age was 57.0 years (range=20-87 years), median number of prior chemotherapy regimens for advanced disease was 3 (range=1-15). T-DM1 was administered for a median of 3 doses (range=1-9) (Table I).
Primary outcome. The adverse events of special interest are summarized in Table II. The most commonly reported adverse event of special interest of any grade was thrombocytopenia (162 patients; 69.8%), followed by hepatotoxicity (140 patients; 60.3%) and peripheral neuropathy (18 patients; 7.8%). The most commonly reported grade ≥3 adverse event of special interest was thrombocytopenia (69 patients; 29.7%), followed by hepatotoxicity (26 patients; 11.2%). NRH was not reported in any patients during the study. LVEF <40% was reported in 1 patient; the LVEF was decreased from 54% at baseline to 30% after the third dose of T-DM1.
Secondary outcome. Overview of adverse events is summarized in Table III. Adverse events were reported in 228 patients (98.3%), grade ≥3 adverse events were reported in 109 patients (47.0%), and serious adverse events were reported in 20 patients (8.6%); no patients died due to adverse events. Overall, the most reported adverse event of any grade was thrombocytopenia (162 patients; 69.8%), followed by AST increased (105 patients; 45.3%), ALT increased (68 patients; 29.3%), and nausea (58 patients; 25.0%). The most reported grade ≥3 adverse event was thrombocytopenia (69 patients; 29.7%), followed by AST increased (12 patients; 5.2%) and hepatic function abnormal (12 patients; 5.2%). The most reported serious adverse events (reported in at least 2 patients) were nausea (3 patients; 1.3%) and pyrexia (3 patients; 1.3%), followed by malaise (2 patients; 0.9%), hepatic function abnormal (2 patients; 0.9%) and anemia (2 patients; 0.9%). Adverse events leading to discontinuation of T-DM1 administration—congestive heart failure, ventricular fibrillation, cellulitis, pneumonitis, and haemorrhagic cystitis—were reported in 5 patients (2.2%). A total of 9 patients required dose modification of T-DM1; the most common adverse event requiring dose modification was decreased platelet count (6 patients; 2.6%). The mean changes in platelet count and levels of AST and ALT among the safety population are shown in Figure 2. During cycles 1-3, the platelet count transiently decreased on day 8 of each cycle (Figure 2A), while AST levels were transiently increased on day 8 of each cycle (Figure 2B) and ALT levels were transiently increased on day 8 of cycle 1 (Figure 2C). The platelet count reached nadir and AST/ALT levels reached a peak with the largest fluctuation range on day 8 of cycle 1.
Baseline characteristics and trastuzumab emtansine exposure among the safety population.
Adverse events of special interest.
Overview of adverse events.

Changes in platelet count and levels of AST and ALT among the safety population (N=232). (A) Changes in platelet count, (B) changes in AST, and (C) changes in ALT.
Exploratory analyses. Grade 4 thrombocytopenia was reported in 9 patients on day 8 of cycle 1. Among them, 7 were permitted to receive T-DM1 with no dose reduction in cycle 2. In 1 patient (shown by a black line in Figure 3A), the platelet count decreased more on day 8 of cycle 2 than on day 8 of cycle 1. Grade ≥3 AST increased was reported in 23 patients on day 8 of cycle 1; 15 of these patients were permitted to receive T-DM1 with no dose reduction in cycle 2. In 1 patient (shown by a black line in Figure 3B), AST level increased after day 1 of cycle 1. Grade ≥3 ALT increased was reported in 4 patients on day 8 of cycle 1; however, all 4 of these patients were permitted to receive T-DM1 with no dose reduction in cycle 2. The ALT level was transiently increased on day 8 of each cycle (Figure 3C).

{kind=link}
{kind=link}
{kind=link}
Changes in platelet count and levels of AST and ALT among patients who were permitted to receive full dose of T-DM1 (3.6 kg/kg) on day 1 of cycle 2, even though these patients had met criteria for modification of administration dosage and schedule of T-DM1 stated in the previously conducted phase II study JO22997. (A) Platelet count in patients who had thrombocytopenia of grade ≥4 on day 8 of cycle 1 and grade ≤2 on day 1 of cycle 2 (n=7). (B) AST levels in patients who had AST increased of grade ≥3 on day 8 of cycle 1 and grade ≤2 on day 1 of cycle 2 (n=15). (C) ALT levels in patients who had ALT increased of grade ≥3 on day 8 of cycle 1 and grade ≤2 on day 1 of cycle 2 (n=4).
To explore the relationship between the severity of thrombocytopenia and haemorrhage, the incidence of those adverse events by grade were cross-tabulated (Table IV). Among 77 patients who had grade 3/4 thrombocytopenia, only 1 patient (1.3%) experienced grade 4 haemorrhage. In this patient, the grade 4 thrombocytopenia was reported on day 8 of cycle 1, while grade 4 haemorrhage (haemorrhagic cystitis) was reported on day 21 of cycle 2 (platelet count of this patient was >300×109/L on day 1 of cycle 3, measured soon after the incidence of grade 4 haemorrhage).
Cross-tabulation of platelet count and haemorrhage.
Discussion
The present phase II study was conducted to evaluate the safety of T-DM1 in Japanese patients with previously treated HER2-positive advanced breast cancer under real-world settings. The administration of T-DM1 was modified according to the laboratory values on day 1 of each cycle. There were no patients who discontinued T-DM1 therapy due to thrombocytopenia or AST/ALT increased. Adverse events of any grade were reported in most patients (98.3%); all reported adverse events were expected and no new adverse events were reported.
The methods of administration dosage or schedule modification of T-DM1 varied between the previous phase II study JO22997 (10) and the present study. In JO22997, the administration dosage or schedule of T-DM1 was modified based on the laboratory values during any time point in the study period because the most abnormal laboratory values during the cycle was considered to be critical for safety. On the other hand, in the present study, the administration dosage or schedule of T-DM1 was modified according to the laboratory values on the administration day based on the pooled safety analysis data (14). Adverse events were reported in 228 patients (98.3%), but only 5 patients (2.2%) discontinued T-DM1; thus, T-DM1 was considered to be well tolerated, with the caveat that the median number of T-DM1 cycles was only 3 in the present study. The incidence and severity of thrombocytopenia observed in the present study were comparable to those in JO22997 (10). The incidence of grade ≥3 thrombocytopenia was 29.7% in the present study compared to 22% in JO22997. In the present study, grade ≥3 AST/ALT increased was 5.2% and 1.3%, respectively, compared to 14% and 8%, respectively, in JO22997.
In this study, the platelet count tended to decrease while the AST/ALT levels tended to increase on day 8 of each cycle and sufficiently recovered to allow continuation of T-DM1 on day 1 of the next cycle. A similar pattern was observed in non-Japanese populations (14). According to the laboratory tests on day 1 of cycle 2, 7 patients who had grade ≥4 thrombocytopenia, 15 patients who had grade ≥3 AST increased, and 4 patients who had grade ≥3 ALT increased on day 8 of cycle 1 were permitted to receive full dose of T-DM1 on day 1 of cycle 2. Although the platelet count, as well as AST/ALT levels, might not have been fully reversible to baseline levels in some patients, they could be recovered to allow continuation of T-DM1 administration. Our investigation on the relationship between incidence of thrombocytopenia and haemorrhage revealed limited association of grade 3/4 thrombocytopenia with grade 3/4 haemorrhage. That is, of the 77 patients who had grade 3/4 thrombocytopenia, only 1 patient (1.3%) experienced grade 4 haemorrhage; in this patient, the grade 4 haemorrhagic event was observed approximately five weeks after grade 4 thrombocytopenia was reported – when the platelet count recovered to the normal level. Thus, the incidence of grade 4 thrombocytopenia was not considered to be associated with grade 4 haemorrhagic event in this patient. A similar pattern was observed in non-Japanese populations. The recently reported pooled safety analysis demonstrated that grade 3/4 thrombocytopenia was rarely associated with grade 3/4 haemorrhage; the proportion of patients who had both grade 3/4 thrombocytopenia and grade 3/4 haemorrhage was 0.67% (6 of 884 patients) (14).
There are some limitations for this study and caution is needed in the interpretation of the results. The present study was exploratory to investigate the safety of T-DM1, without any comparisons. Furthermore, the median number of cycles was relatively small.
In conclusion, our safety analyses demonstrated that T-DM1 was well tolerated within the early phase of T-DM1 therapy in Japanese patients with HER2-positive advanced breast cancer who were previously treated with a trastuzumab-containing regimen. No new safety signals were observed.
Acknowledgements
Authors would like to express our deepest gratitude to the patients, in addition to the investigators and staff at the study sites for their contribution to the study.
The present study was sponsored by Chugai Pharmaceutical, Co., Ltd. (Tokyo, Japan). Medical writing service was provided by Koki Yamashita, PhD of Maxwell International, Inc. (Chiba, Japan), which was funded by Chugai Pharmaceutical Co., Ltd. All the Authors contributed to revision of the drafts, approved the final version, and made the decision to submit the manuscript for publication.
Footnotes
This article is freely accessible online.
Conflicts of Interest
Junichiro Watanabe reports receiving manuscript fees from Chugai Pharmaceutical Co., Ltd., honoraria from AstraZeneca K.K., Chugai Pharmaceutical Co., Ltd., Eisai Co., Ltd., Kyowa Hakko Kirin Co., Ltd., Novartis Pharma K.K., Taiho Pharmaceutical Co., Ltd., and being an advisory board member of AstraZeneca K.K., and Eisai Co., Ltd.; Yoshinori Ito reports receiving manuscript fees from Chugai Pharmaceutical Co., Ltd., Eisai Co., Ltd., Novartis Pharma K.K., and Taiho Pharmaceutical Co., Ltd., and research funds from Chugai Pharmaceutical Co., Ltd., Parexel International Inc., Novartis Pharma K.K., AstraZeneca K.K., Taiho Pharmaceutical Co., Ltd., Eisai Co., Ltd., Daiichi Sankyo Co., Ltd., and EPS Co.; Norikazu Masuda reports receiving honoraria from Chugai Pharmaceutical Co., Ltd., Kyowa Hakko Kirin Co., Ltd., and AstraZeneca K.K.; Toshimi Takano reports receiving research funds from Chugai Pharmaceutical Co., Ltd.; Shintaro Nakagawa and Kazumitsu Kanatani report being employees of Chugai Pharmaceutical, Co., Ltd.; Takahiro Nakayama reports receiving honoraria from Chugai Pharmaceutical Co., Ltd., Novartis Pharma K.K., and AstraZeneca K.K.; Toshiaki Saeki, Shintaro Takao, Kazuhiko Nakagami, and Koichiro Tsugawa report no conflict of interest.
- Received February 27, 2017.
- Revision received March 21, 2017.
- Accepted March 22, 2017.
- Copyright© 2017, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved