

Abstract
Background/Aim: To study the prevention of chemotherapy resistance, we have previously designed models of drug-resistant ovarian cancer. We here report an in vivo model of cisplatin-resistant small cell lung cancer (SCLC). Materials and Methods: Mice bearing H526 SCLC xenografts received intraperitoneal pretreatment with a sub-effective cisplatin dose (0.75-1.5 mg/kg) or no pretreatment (controls). Seven days later, all mice received a higher cisplatin dose (3.0 mg/kg), and tumor response was recorded. Cell cultures initiated from pretreated and control xenografts were tested for cisplatin resistance and for glutathione-S-transferase (GST) activity. Results: Pretreatment with 1.5 mg/kg cisplatin induced resistance to 3.0 mg/kg cisplatin. Cells from a pretreated tumor were cisplatin resistant and had nearly twice the GST activity as cells from a control tumor. Conclusion: Such cells may prove useful for identifying other resistance mechanisms and thus guide the selection of potential preventative agents to be tested in the in vivo model.
- Cisplatin
- drug resistance
- SCLC
Cisplatin is the treatment of choice for cancer such as ovarian and small cell lung cancer (SCLC). However in these types of cancer, cisplatin chemotherapy often becomes ineffective due to development of resistance (1, 2). Many investigations of drug resistance have been performed on cells or tumors already resistant to chemotherapy (1-3). In some cases, these studies have identified potential causes of resistance. For example, numerous studies have described an increase in glutathione-S-transferase (GST) in cisplatin-resistant ovarian cancer and SCLC (1-3). Resistant tumors and cells have also been used to investigate treatment methods that increase tumor cell sensitivity or reverse drug resistance. However, treatments designed to overwhelm or reverse a tumor's resistance mechanisms are likely to also weaken the patient's normal cells. Thus the clinical success of these treatments may be limited by their severe toxicity (1-3). In contrast, an agent capable of preventing tumor cells from developing resistance need not weaken normal cells and thus may be better tolerated by patients (4). However this prevention strategy poses special challenges compared to reversal studies.
In order to test an agent's ability to prevent resistance, it is necessary to identify a time by which resistance would have predictably developed. We previously designed models of ovarian cancer in which resistance occurred within 7 days of initial treatment (4, 5). Using these models, we found that the selenium compound selenite prevents the development of resistance to platinum-based chemotherapy (6-8). We also examined causes of resistance in ovarian cancer models, and reported an increase in glutathione or its related enzyme GST in these ovarian tumor xenografts and cells as they develop resistance (4, 9).
The most lethal form of lung cancer is SCLC, which rapidly develops resistance to chemotherapy (10). Numerous studies have examined chemotherapy resistance in SCLC cells and tumors (11-20), including the reversal of resistance (19, 20). However to our knowledge, none have focused on its prevention. We designed an in vivo model of the development of resistance to cisplatin in mice bearing SCLC xenografts. Cells from these xenografts, tested in vitro, were found to reflect the relative resistance of the tumor from which they were derived. We also tested cells from these xenografts for GST activity, a known mechanism of resistance in SCLC (18, 19), and found that cells from the resistant xenograft have significantly higher GST activity than those from the control xenografts. This evidence supports the use of the in vivo model to test whether agents capable of inhibiting GST activity are effective in preventing the development of cisplatin-resistant SCLC xenografts. In addition, cells derived from the in vivo model may be used to identify other resistance mechanisms and thus guide the selection of additional potential preventative agents.
Materials and Methods
Cell culture. SCLC NCI-H526 Cells American Type Culture Collection (Bethesda, MD, USA) were cultured in RPMI plus 10% Gibco brand fetal bovine serum (FBS) from Thermo-Fisher Scientific Life Sciences, Pittsburgh, PA USA. Cells were maintained at 37°C and 5% CO2 and were transferred into fresh medium to maintain approximately 50-75% confluence.
Animal protocol. All experiments involving mice were approved by Rutgers University Animal Welfare Committee protocol #11-012 and were carried out under the supervision of University veterinarians. Female athymic nude mice (Harlan, Indianapolis, IN, USA) were housed in sterile microisolator cages. At 5-6 weeks they were inoculated subcutaneously in the flank with 0.1 ml of cell suspension containing 2×106 SCLC NCI-H526 cells from the cultures described above. Visible tumors appeared in approximately 2 weeks. Thereafter, tumor size was measured daily with calipers, and volume was calculated using the formula: volume= length×width2/2.
Tumor growth assays. After approximately 15 days, when tumors were 0.8 to 1.2 cm3, replicate mice received a ‘pretreatment’ dose (0, 0.75 or 1.5 mg/kg) of cisplatin (Sigma-Aldrich, St. Louis, MO, USA) on either the third, fifth or seventh day before the test dose (3.0 mg/kg). Others received no pretreatment and served as controls. Mice were sacrificed 3 to 5 days after receiving the high dose.
Deriving cells from xenografts. Tumor-bearing mice receiving either no treatment or a single i.p. pretreatment with cisplatin (1.5 mg/kg) were sacrificed on day 15 (without receiving high-dose treatment). Tumors were removed and fragments were homogenized in culture medium containing Penn/Strep and 15% FBS. The cell suspension was seeded into culture flasks and incubated for 2 weeks. Cells were removed from the flasks, centrifuged, counted and tested for resistance and GST activity.
Relative cisplatin-sensitivity assay. Cells were seeded in replicate culture dishes at 1×104 cells/ml. After 48 h, cells were pelleted and resuspended in fresh medium with a cisplatin concentration of either 0, 10 or 20 μM. After 24 h, cells were counted, the mean±SEM were calculated and values expressed as the percentage that of non-treated cells. Two-tailed t-test analysis was performed by GraphPad Prism (LaJolla, CA, USA) to evaluate whether responses differed significantly.
GST assay. Cells were counted and extracts prepared following the method of Habig and Jakoby (21). GST activity on the conjugation of 1-chloro,2,4-dinitrobenzene with reduced glutathione (GSH), was measured by the absorbance at 340 nm. The GST activity/ml of the extract was calculated as: (Δ absorbance/min) ×0.0096 μM−1/cm ×0.3 ml. The number of cells per milliliter of extract was used to calculate the GST activity per cell. Two-tailed t-test analysis was performed by GraphPad Prism to evaluate whether responses differed significantly.

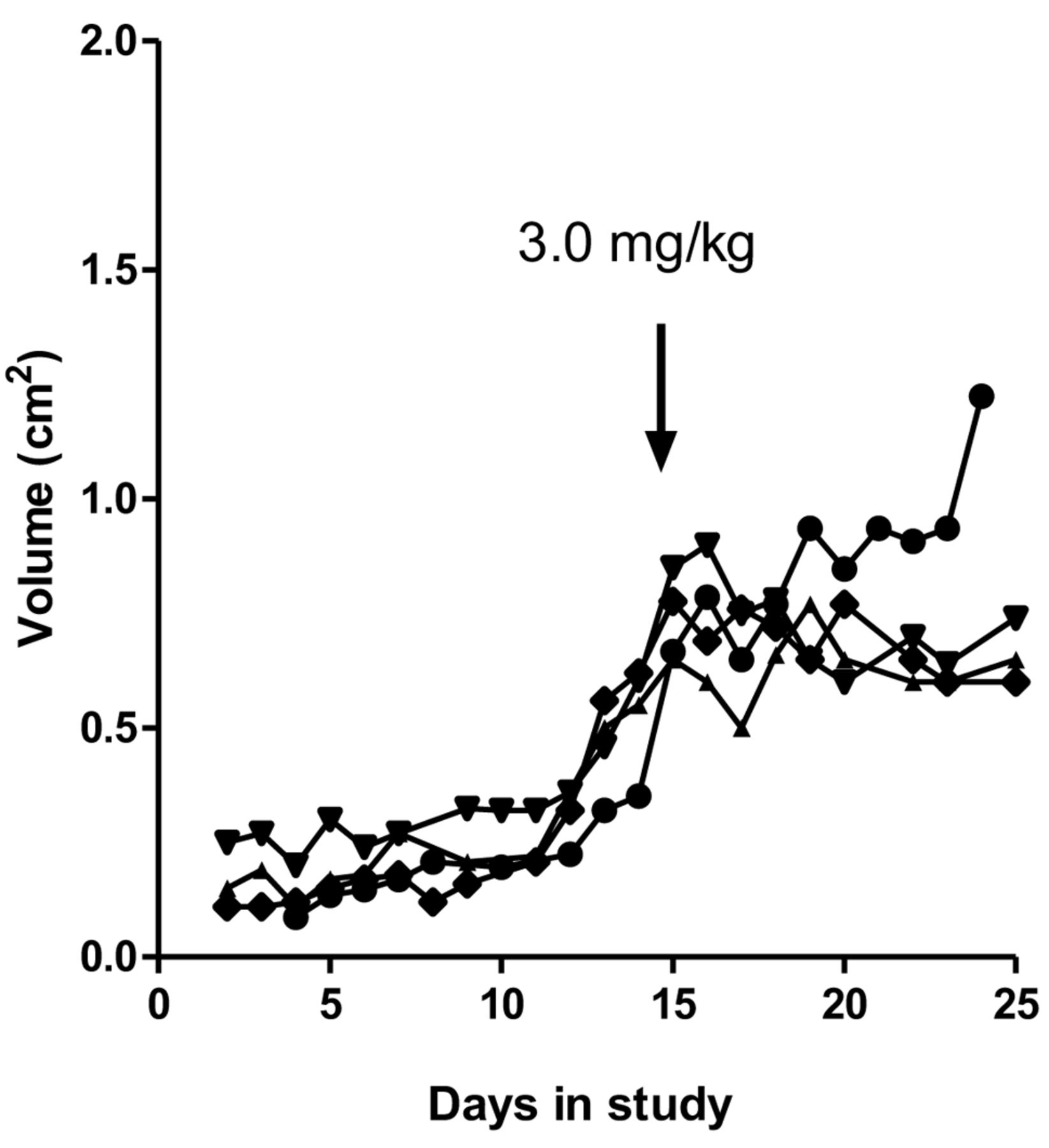
Effect of high-dose cisplatin on xenografts receiving no pretreatment. Mice bearing NCI-H526 SCLC xenografts were treated with 3.0 mg/kg cisplatin on the indicated day in the study. Each curve represents the volume of an individual mouse xenograft.
Results
As in our ovarian cancer model of development of cisplatin resistance, our SCLC model design included exposing xenografts to ‘pretreatment’ with sub-effective cisplatin, followed by a single, high, effective dose of cisplatin after a specific time interval. A positive result (i.e. development of resistance) was indicated by the continuation of exponential tumor growth following this high dose. We tested a range of potential effective doses and determined that treatment with 3.0 mg/kg cisplatin resulted in a cessation of exponential growth for at least 3 days in mice receiving no pretreatment (Figure 1). Lower doses had little or no effect, except for tumors smaller than 0.65 cm3 (not shown). Accordingly, we chose 3.0 mg/kg as the high dose and excluded tumors smaller than 0.65 cm3 at the time of the high dose treatment from the study.
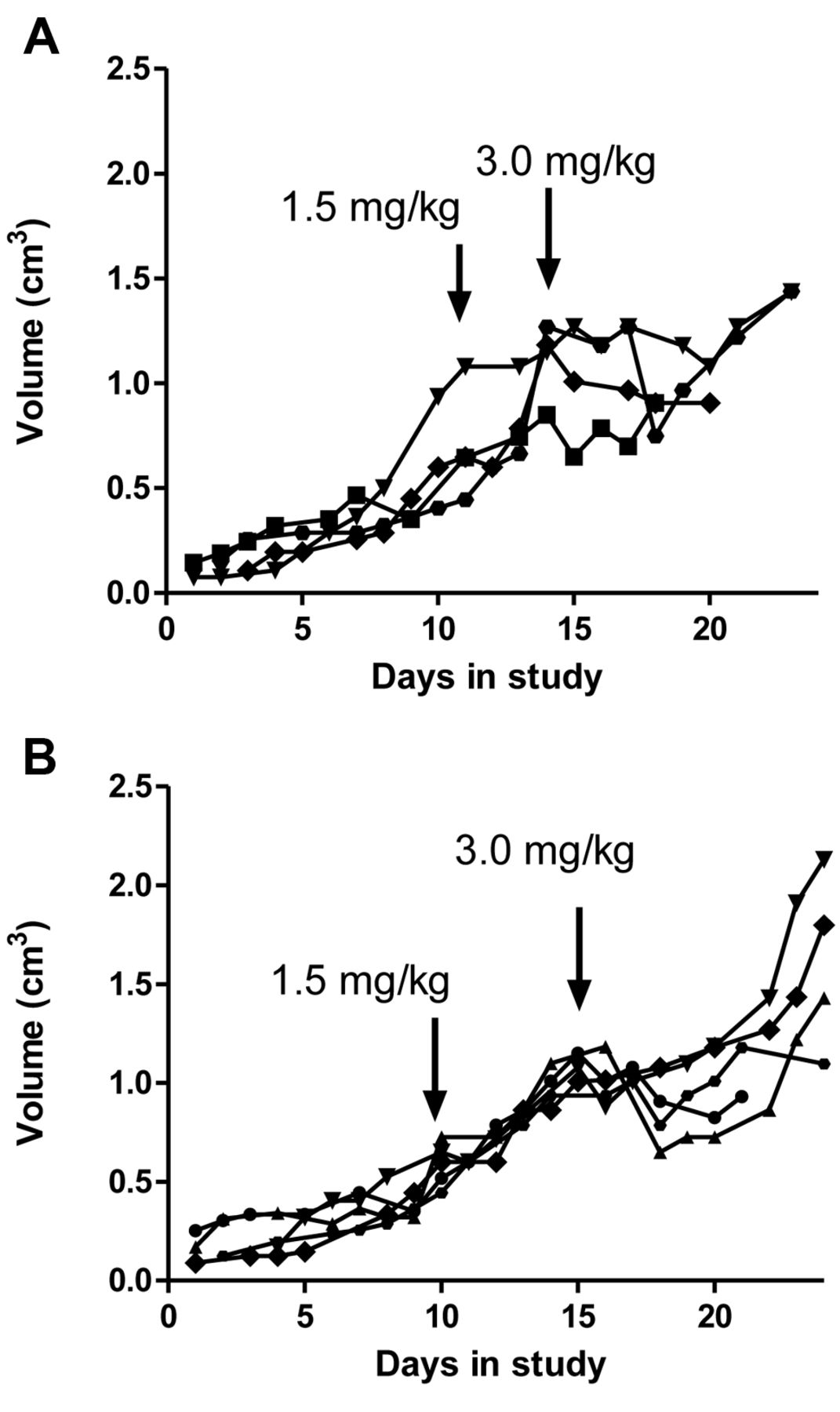
In order to permit the observation of resistance, tumor growth must be unhindered by the pretreatment dose of cisplatin. We tested a range of pretreatment doses, administered at different times before the high test dose. In our ovarian cancer model, the time interval between the pretreatment and test dose was 7 days. Thus our initial experiments included pretreatment 7 days prior to the administration of high-dose cisplatin. Figure 2 illustrates two pretreatment doses which resulted in the development of resistance to the high dose after 7 days (Figure 2). Pretreatment with 1.5 mg/kg virtually eliminated the inhibitory effect of the high-dose cisplatin on tumor growth (Figure 2A) compared to the response of non-pretreated controls (Figure 1). Thus, 1.5 mg/kg pretreatment induced resistance in these SCLC xenografts. Resistance was also evident following pretreatment with a lower cisplatin dose of 0.75 mg/kg (Figure 2B).

Effect of different pretreatment doses on response to high-dose cisplatin administered 7 days later. Mice bearing tumors were treated with 3.0 mg/kg cisplatin on the indicated day. A: Pretreatment with 0.75 mg/kg cisplatin. B: Pretreatment with 1.5 mg/kg cisplatin. Each curve represents the volume of an individual mouse xenograft.

Effect of different time interval between pretreatment and high-dose cisplatin treatment. Mice bearing tumors were treated with 3.0 mg/kg cisplatin on the indicated day in the study. Pretreatment 3 days (A) and 5 days (B) before high-dose cisplatin treatment. Each curve represents the tumor volume of an individual mouse xenograft.
In order to establish the optimum conditions for resistance development, we varied the time intervals between pretreatment and high-dose treatment. The effects of 1.5 mg/kg pretreatment, administered either 3 or 5 days prior to the test dose, are shown in Figure 3. Neither of these time intervals resulted in the degree of resistance observed using the 7-day interval. Thus the most successful model included a resistance-inducing pretreatment with 1.5 mg/kg cisplatin administered 7 days before the high dose of 3.0 mg/kg, as shown in Figure 2A.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
A: Effect of in vitro cisplatin on C-H526 cells from control xenograft. Responses to each of the in vitro cisplatin doses were significantly different from each other and from cells receiving no in vitro cisplatin (p<0.005). B: Effect of cisplatin in vitro on R-H526 cells from pretreated (resistant) xenograft. There was no significant effect of either cisplatin dose on cell survival (p>0.05) showing that resistant and sensitive phenotypes are maintained in vitro in cells from tumors.
Xenografts were removed from replicate mice on day 7 following pretreatment, before treatment with high-dose cisplatin, to test whether this resistance persists ex vivo. Tumors were diced and cells grown in cultures for two weeks. Cells were then exposed to cisplatin and the number of cells was determined. We found that cells from a xenograft pretreated with 1.5 mg/kg cisplatin (designated R-H526) exhibited resistance to 10 or 20 μM cisplatin in vitro compared to cells from a control xenograft (C-H526) (Figure 4). Thus the tumor cells retained their resistant or sensitive phenotype in vitro.
We investigated whether GST activity had been influenced by cisplatin pretreatment in our model, and found that cells from the resistant xenograft R-H526 exhibited 1.04±0.17 activity units per cell compared to 0.52±0.11 activity units per cell in the C-H526 (sensitive) xenograft. A two tailed t-test confirmed that these means and standard errors from replicate experiments were statistically different (p<0.001).
Discussion
A major trend in cancer research focuses on the activity of natural products and dietary factors reported to reduce cancer incidence (22-25). However, in contrast to the low dietary levels associated with cancer prevention, the doses of such substances required to induce death of cancer cells may not be achievable in patients without producing toxic side-effects (26, 27). In contrast, the prevention of resistance may be achieved by much lower, better-tolerated doses. For example, using a xenograft model of the development of resistance in ovarian cancer (6, 7), we found that selenite prevents resistance at a dose which is an order of magnitude lower than that required for inhibition of tumor growth and tumor cell proliferation (28). In future studies, we will use this new SCLC model to screen potential resistance-preventative agents.
Glutathione and related enzyme GST have been associated with drug resistance in many types of cancer. In our previous model of resistance development by ovarian cancer cells, we found that GST levels were elevated in cells in which resistance had developed (29). Numerous studies in both non-SCLC and SCLC cells have described the overexpression of GST as a cause of resistance to chemotherapy (10, 30, 31). Novel strategies designed to lower the expression of GST continue to be explored in an effort to identify methods of reversing cisplatin resistance. For example, deregulation of miRNAs has been shown to sensitize human lung adenocarcinoma cells to cisplatin by targeting GSTP1 (19). Others have reported that overexpression of FAS reversed cisplatin resistance by targeting GSTP expression (20). The data on cells taken from SCLC xenografts indicate elevated GST levels in cells from xenografts that had developed cisplatin resistance. Using cells from our ovarian cancer model, we found that selenite prevented the increase in GST levels at the doses at which it prevented resistance (28). We plan to test whether selenite or other GST inhibitors can prevent the development of cisplatin resistance in our SCLC model.
Acknowledgements
This study was funded by a grant from the Pennsylvania State System of Higher Education.
Footnotes
This article is freely accessible online.
- Received August 23, 2016.
- Revision received October 23, 2016.
- Accepted October 24, 2016.
- Copyright © 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved