

Abstract
Background: Aberrant histone deacetylase expression may cause imbalance between acetylation and deacetylation of histone and play roles in tumor transformation. We found that histone 3 was modulated in human hepatocellular carcinoma. We determined if histone 3 modulation is related to the aberrant expression of histone deacetylase. Materials and Methods: We analyzed human liver and hepatocellular carcinoma tissues and fibroblast and fibrosarcoma cell lines for the expression of histone 3, histone deacetylase 1 and acetylated histone 3 using immunohistochemistry, western blot and immunofluorescence. Results: Histone deacetylase 1 and histone 3 were more strongly detected in hepatocellular carcinoma tissue and fibrosarcoma cells than in liver tissues and fibroblast cells, respectively. However, acetylated histone 3 was more strongly expressed in normal liver and fibroblast cells and less expressed in hepatocellular carcinoma and fibrosarcoma cells. Conclusion: Histone deacetylase 1 overexpression and hypoacetylation of histone 3 might play critical roles in the modulation of histone 3 in human hepatocellular carcinoma.
- Acetylation
- cytokeratin
- hepatocellular carcinoma
- histone 3
- histone deacetylase
- modulation
In the eukaryotic nucleus, the organization and packaging of DNA are achieved by the addition of histone proteins to form chromatin. Post-translational modifications of histones, such as acetylation, phosphorylation, methylation and ADP-ribosylation, frequently alter their function (1). Alterations in chromatin structure play a central role in the regulation of gene transcription, and recent studies suggest that these alterations may be important in the process of neoplasia (2).
The acetylation status of histones alters the chromatin structure, which, in turn, is involved in gene expression. Histone hyperacetylation correlates with transcriptional activity of the genome, whereas hypoacetylated histones are associated with silent genes (3). Two classes of enzyme affect the acetylation status of histones: histone acetyltransferases (HATs) and histone deacetylases (HDACs) (4). Alterations in the activities of HAT and HDAC, as well as aberrant acetylation or deacetylation of histone, lead to disorders including cancer (5). HDACs catalyze histone deacetylation and repress transcription of specific genes, such as p21, resulting in cell-cycle activation and cell proliferation. HDAC inhibitors, such as butyrate, inhibit HDAC activity and activate the p21 gene, which in turn causes the arrest of cancer cell growth (2). These studies suggest that HDACs have a role in human tumorigenesis and that HDAC inhibitors might be a good choice for cancer therapy. Although some HDAC inhibitors have been utilized in cancer therapy, the role of HDACs in tumor transformation has not been investigated.
In our previous study, we found proteins (12.4-18.4 kDa in molecular weight) subsequently identified as histone 3 (H3) that co-immunoprecipitated with cytokeratin 18 (CK18) in human hepatocellular carcinoma (HCC) tissues (6) and in the PLC/PRF/5 hepatoma cell line (7). Modulation of H3 in HCCs was also observed in the study (8). These phenomena of H3 modulation and the co-immunoprecipitation of H3 with CK18 were also seen in transitional cell carcinoma (9) and other epithelial tumors (10). These data suggest that H3 modulation might play important roles in the process of tumor transformation, including human HCC.
Recently, we also found that H3 modulation resulted in nuclear instability of human HCC cells (11). We propose that aberrant acetylation of H3 in human HCC might be responsible for this phenomenon. We designed this study to investigate the expression of H3, HDAC1 and acetylated histone 3 (AH3) in human HCC and normal liver tissues using immunohistochemistry and western blot analysis. In vitro experiments using cell lines of normal fibroblast cells (HS68) and a fibrosarcoma (HT1080) cell line were performed by western blot and immunoflurescence to confirm the results found in tissues. We hoped to clarify whether H3 modulation in human HCC is related to the acetylation status of histone.
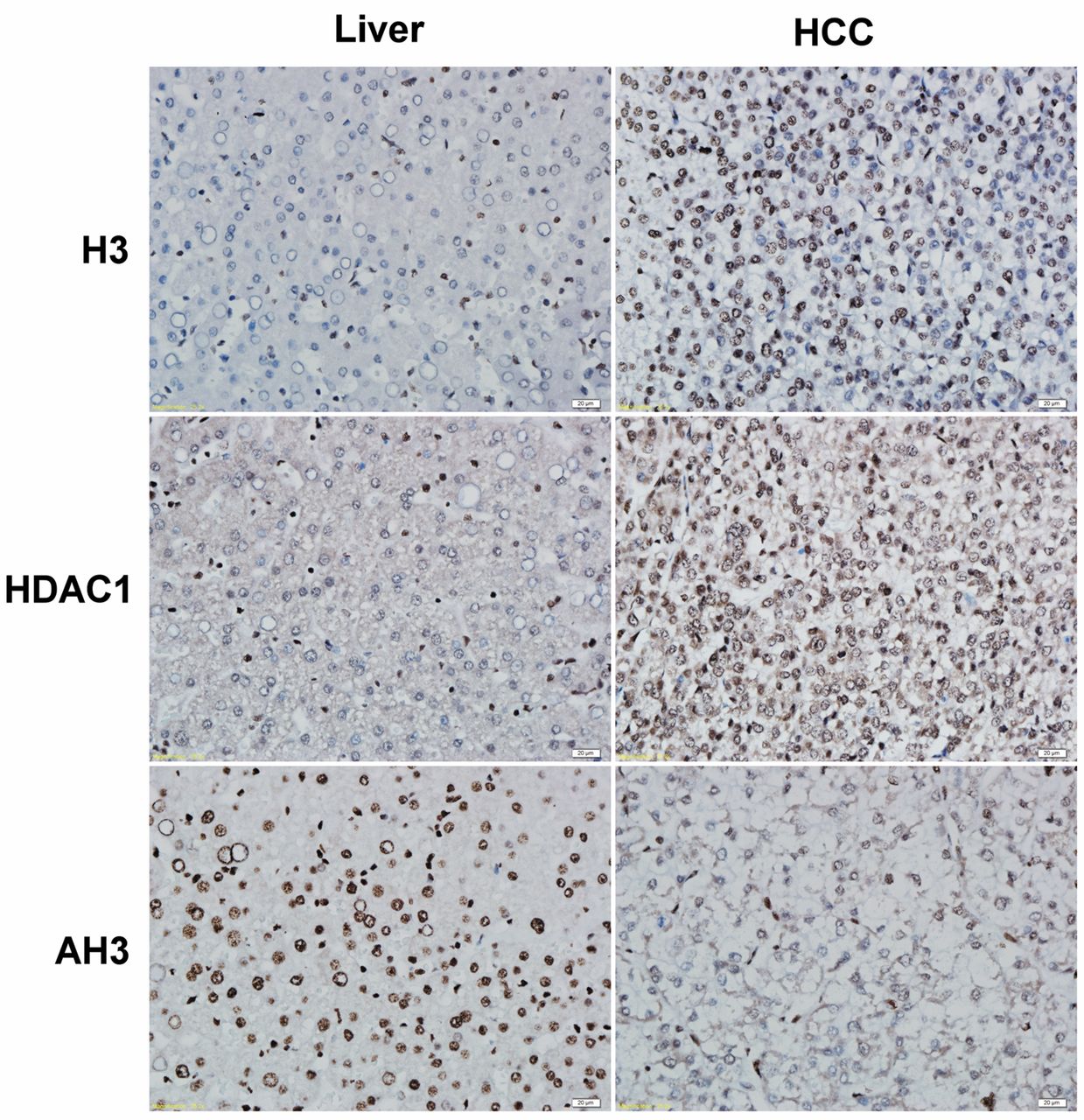
Immunohistochemical staining of histone 3 (H3), histone deacetylase 1 (HDAC1) and acetylated histone 3 (AH3) in human hepatocellular carcinoma (HCC) and liver tissues. The tumoral part of the HCC section revealed strong nuclear staining for H3 and HDAC1, while the non-tumoral part stained weakly for H3 and HDAC1. AH3 staining was positive n the non-tumoral part of the HCC section, but the tumoral part exhibited practically negative staining. HCC: Tumoral part of HCC section, Liver: non-tumoral part of HCC section; original magnification ×400.
Materials and Methods
Tissue samples and antibodies. Five cases of freshly surgically resected tissue from patients with primary HCC, as well as their accompanying paraffin blocks, and one fresh specimen from a case with liver trauma obtained by surgical resection, without any neo-adjuvant therapy at the Department of Surgery, Chang Bing Show Chwan Memorial Hospital, Changhua County, Taiwan, ROC. One case of HCC was histopathologically-diagnosed as grade II-III and the others were grade II. The fresh samples were stored at −80°C before protein extraction. The following commercial primary and secondary antibodies were used for immunohistochemistry, western blot and immunofluorescence assay: anti-H3 and anti-AH3 (Cell Signaling Technology, Boston, MA, USA) and anti-HDAC1 (Abcam Inc., Cambridge, MA, USA) were polyclonal primary antibodies; anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) was conjugated to biotin for immunohistochemistry, horseradish peroxidase for western blot analysis and rhodamine for immunofluorescent staining.

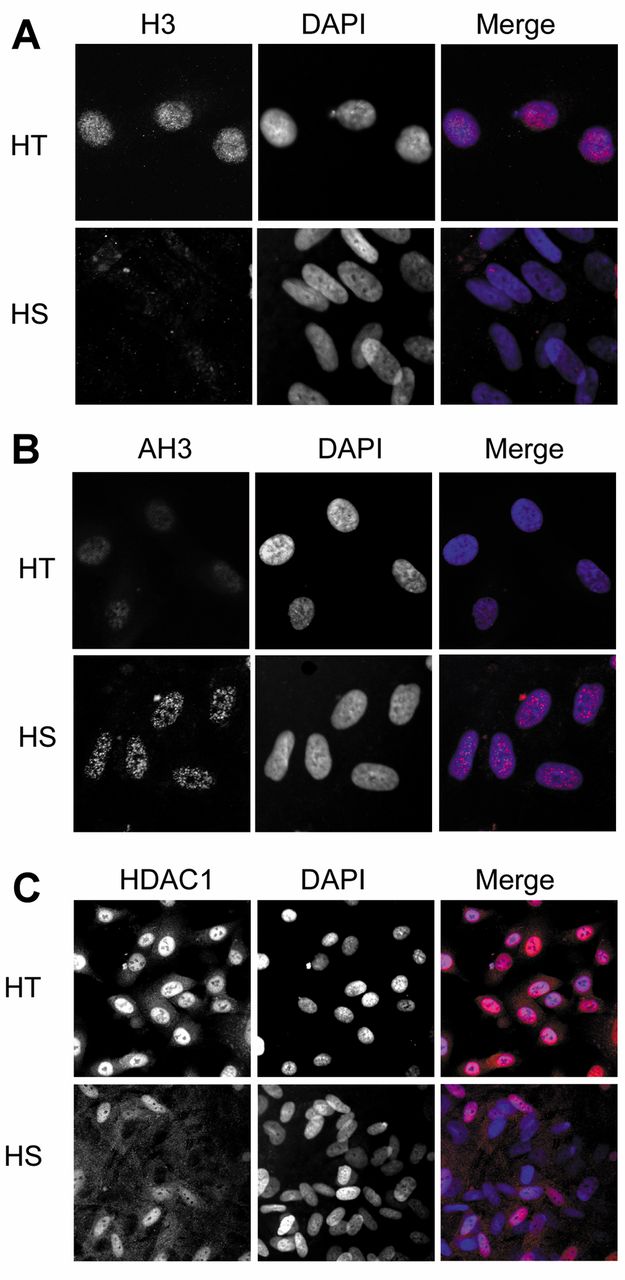
Immunofluorescence assay of histone 3 (H3), histone deacetylase 1 (HDAC1) and acetylated histone 3 (AH3) in HS68 cells and HT1080 cells. A: H3 was mainly distributed in the nuclei of HT1080 cells, while the HS68 cells exhibited weak staining. B: HDAC1 was more abundant in the nuclei of HT1080 cells than in the nuclei of HS68 cells. C: AH3 in HS68 cells was stronger when compared to HT1080 cells. Original magnification, ×1,000. DAPI (4’,6-diamidino-2-phenylindole).
{kind=link}
{kind=link}
{kind=link}
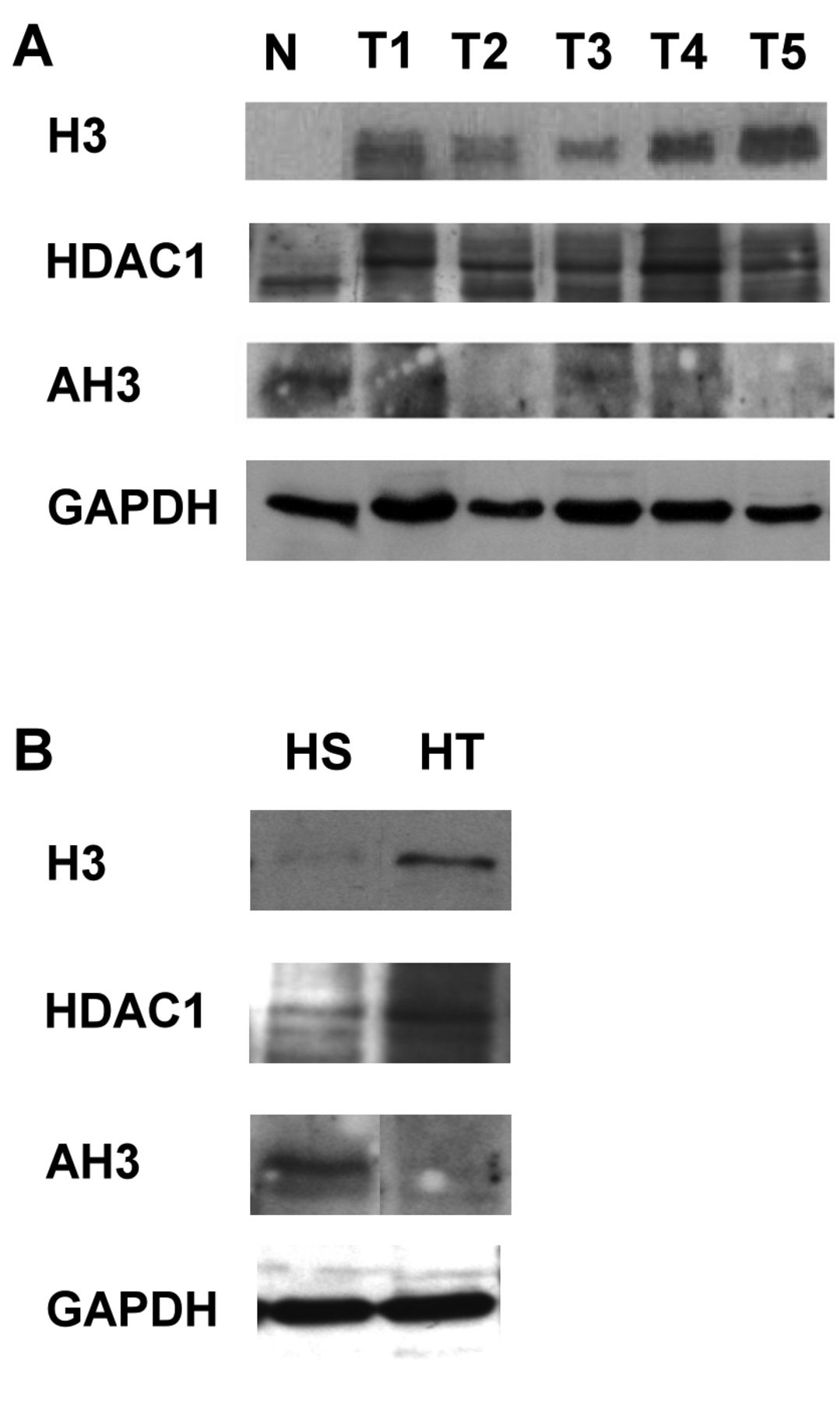
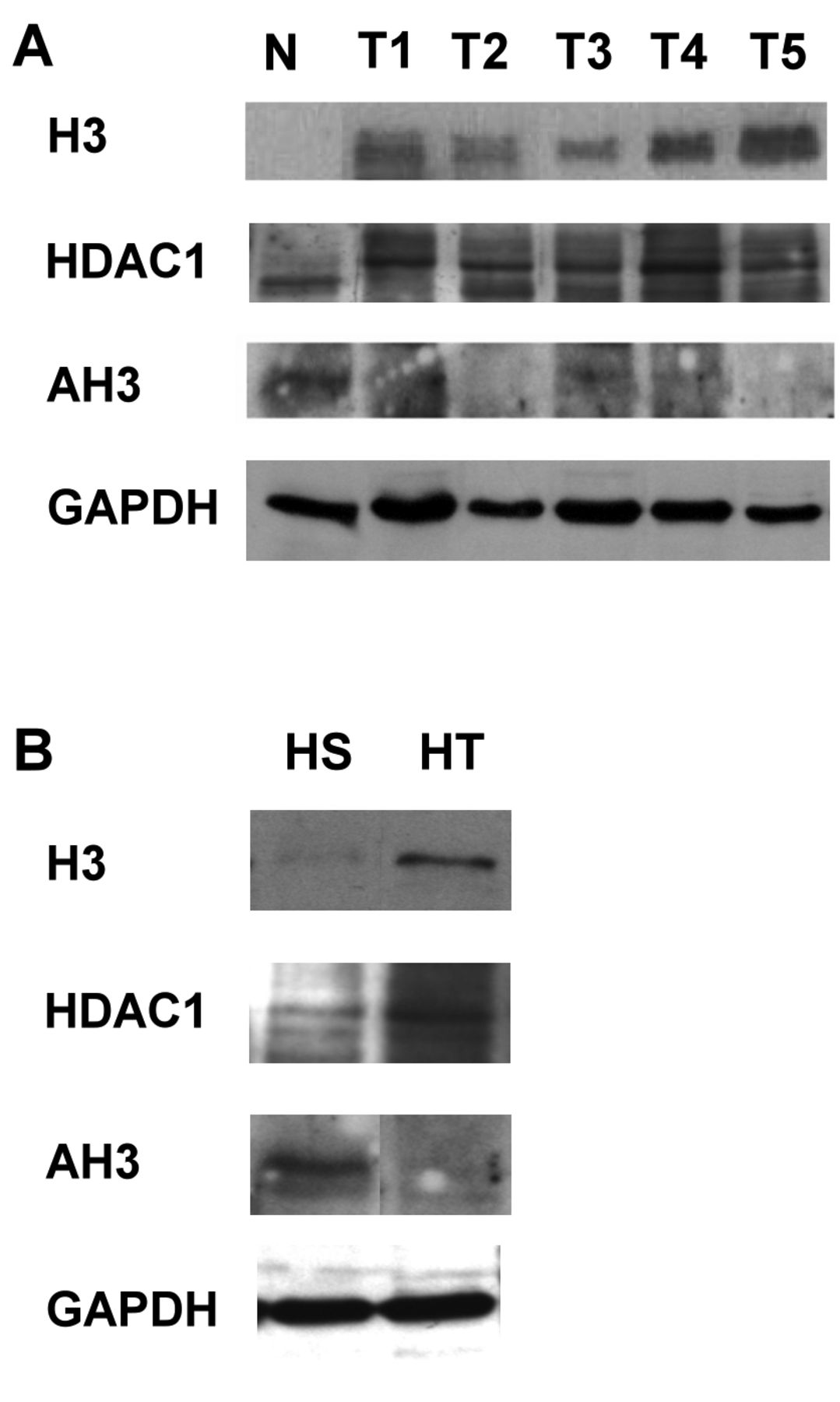
Western blot analysis of hepatocellular carcinoma tissues and cell lines. Total proteins extracted from normal liver (N) and HCC (T1-5) tissues were analyzed. The expression of histone 3 (H3) and histone deacetylase 1 (HDAC1) showed up-regulation in HCC tissues (A) and HT1080 cells (B) compared to normal liver tissues and HS68 cells. Normal liver tissue and HS68 cells exhibited positive staining for AH3, while in HCC tissues and HT1080, AH3 was down-regulated.
Cell culture. Normal human fibroblast cells (HS68 cells) and cells of fibrosarcoma (HT1080 cells) from the American Type Culture Collection (Rockville, MD, USA) number CRL-1635™ and CCL-121™, were grown in Dulbecco's minimum essential medium supplemented with 10% fetal bovine serum, 50 units/ml penicillin and streptomycin and cultured 2-4 days at 37°C in 5% CO2. The medium was replaced every two days.
Total protein extraction from HCC tissues and HS68 and HT1080 cells. Total protein was extracted from tissue specimens and cultured cells. Tissue samples (less than 1 g) were minced with shears and homogenized in a lysis buffer containing a protease inhibitor cocktail consisting of 50 mM Tris-HCl (pH 7.6), 150 mM NaCl, 5 mM ethylenediaminetetra-acetic acid, 0.5% Triton X-100 and 0.5% Nonidet P-40. Unbroken cells and connective tissue were removed from the homogenate by centrifugation at 500 × g for 10 minutes at 4°C. Cells adherent to the T25 tissue culture flask were briefly washed with ice-cold phosphate-buffered saline and were scratched into fresh phosphate-buffered saline and then centrifuged at 500 ×g for 5 min. The cell pellet was then dissolved in lysis buffer. Samples were kept on ice at all times and supernatants were stored at −80°C until use.
Western blot. Total protein extracted from tissue specimen and cell lines were mixed with loading dye and heated at 95°C for 5 min. The total lysates were loaded into 10% sodium dodecyl sulfate polyacrylamide gel. After electrophoresis, proteins were transferred to polyvinylidene fluoride membrane by using semi-dry transfer method (Bio-Rad Laboratory Inc., Hercules, California, USA). The membrane was placed in 5% non-fat milk (prepared with phosphate-buffered saline with Tween-20; PBST) and blocked for 1 hour. The membrane was washed by 1×PBST three times for 5 min before adding primary antibody. The dilution of antibodies was as 1:1,000 for anti-H3, 1:2,000 for anti-HDAC1 and 1:2,000 for anti-AH3 in PBST. After 1-hour incubation, the membrane was washed by 1×PBST three times for 5 min then secondary antibody (1:4000 in PBST) was added. The membrane was washed by 1×PBST three times for 5 minutes and then LuminataTM Crescendo Western HRP substrate (Millipore Corporation, Billerica, USA) was added and the membrane photographed by a Chemiluminescence Imaging System (FUJI Photo Film Co., Tokyo, Japan).
Immunofluorescence. HS68 and HT1080 cells were cultured on coverslips in 24-well plates (4×103 cells per well). After 48 hours, the cells were washed with iced phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, and 1.5 mM KH2PO4, pH 7.4) and fixed with 3.7% paraformaldehyde at room temperature for 5 minutes. The cells were then washed three times with phosphate-buffered saline and then treated by 0.1% Triton X-100 for 2 minutes. The cells were stained for 1 hour with primary polyclonal antibodies (1:100 diluted anti-H3, 1:200 diluted anti-HDAC1 and 1:200 diluted anti-AH3) at room temperature and again washed with phosphate-buffered saline before being incubated with rhodamine-conjugated anti-rabbit IgG secondary antibody for 30 min at room temperature. The unbound antibodies were removed by washing twice for 10 min in phosphate-buffered saline, and the fluorescence was evaluated under a fluorescence microscope (Olympus BX51; Olympus, Tokyo, Japan).
Immunohistochemistry. De-paraffinized and rehydrated tissue sections were treated with 3% H2O2 for 10 min to eliminate endogenous peroxidase activity. Nonspecific binding sites were blocked with bovine serum albumin for 10 minutes. The sections were then incubated with polyclonal antibodies raised for H3 (1:50 dilution), HDAC1 (1:200 dilution) and AH3 (1:100 dilution) for 1 h at room temperature. Biotinylated secondary antibody (anti-rabbit IgG,) was added and the final signal was revealed by the avidin-biotin peroxidase technique in the presence of hydrogen peroxide. These sections were evaluated under light microscopy (Olympus BX51, Olympus).
Results
Immunohistochemical analysis of H3, HDAC1 and AH3 in human normal liver and HCC tissues. Five cases of human HCC were processed for immunohistochemistry to observe the H3, HDAC1 and AH3 expression (Figure 1). The patterns of immunohistochemical staining were different in cancerous cells than in non-cancerous liver tissues. The nuclei of HCC cells strongly stained for H3, while liver cells stained weakly. The nuclei of HCC cells also showed relatively strong staining for HDAC1 compared to their non-cancerous counterpart. The staining for AH3 revealed strongly nuclear staining in non-cancerous liver cells but faint or practically negative staining in nuclei of HCC cells.
Expression of H3, HDAC1 and AH3 in cultured cells by immunofluorescnese assay. We localized H3, HDAC1 and AH3 within HS68 (human fibroblast) and HT1080 (fibrosarcoma) cells by immunofluorescence assay. These two cell lines were used as a model to compare the expression of H3, HDAC1 and AH3 between normal and cancerous cells. We chose HS68 and HT1080 cell lines because there is no normal human liver cell line available to contrast with the human HCC cell line. As seen in Figure 2, the nuclei of HS68 cells did not stain strongly by H3 and HDAC1, as was the case for the nuclei of HT1080 cells. However, the immunostaining of AH3 in HS68 cells was stronger when compared to HT1080 cells. These findings suggest that in cancer cells, the expression of HDAC1 is important for the regulation of H3 acetylation status.
Determination of H3, HDAC1 and AH3 levels by western blot analysis in tissues and cell lines. To clarify the above finding, five HCC tissues and normal liver tissue proteins were extracted and subjected to western blot analysis. In Figure 3A, the results show that the expression of H3 was up-regulated in human HCC compared to normal liver tissue. H3 was acetylated in normal tissue and most of the HCC tissue samples did not exhibit H3 acetylation. However, the expression of HDAC1 was higher in HCC tissues than in the normal liver tissue. HDAC1 was also modulated and shown to have higher molecular weight in the western blot. Reduced acetylation of H3 in HCC tissues might therefore be due to the up-regulation of HDAC1.
In vitro data revealed the same condition, the results in Figure 3B show that H3 and HDAC1 were up-regulated in cancer cells (HT), but were scarcely seen in normal cells (HS). In contrast, AH3 was less prominent in cancer cells compared to normal cells. H3 acetylation was reduced in cancer cells, a modification possibly related to the activity of HDAC1.
Discussion
In the present study, we investigated whether H3 modulation in human HCC is related to acetylation status of histone. We used in vitro data and tissue samples to explain this phenomenon. In human HCC tissues, the expression of H3 was modulated and up-regulated, HDAC1 was up-regulated, and AH3 was reduced when compared to normal liver tissue. In vitro data showed the expression of H3 was up-regulated in HT1080 cells; again, we found HDAC1 was up-regulated and AH3 was reduced when compared to HS68 cells. Based on these data, we conclude that the modulation of H3 in human HCC is related to H3 hypoacetylation due to HDAC1 up-regulation.
The investigation of histone in human HCC in our laboratory originated in a study of CK18 in which we found that CK18 was modulated and that it co-immunoprecipitated with H3, which was also modulated, in human HCC (6, 8). We hypothesized that H3 modulation might cause nuclear instability of human HCC cells, which led to it being co-immunoprecipitated with CK18 (11). In this study, we further confirmed, using techniques of immunohistochemistry and western blot, that expression of H3 was up-regulated in human HCC. We also observed that H3 modulation was related to the up-regulation of HDAC1 and the hypoacetylation of H3 in human HCC. We focused on HDAC1 because other research reported that altered expression of HDAC1 in HCC may regulate key events in liver carcinogenesis (12). The roles of other classes of HDACs in human HCC are being investigated.
We also found that in normal liver tissue and HS68 cells, H3 was not seen on western blot assay (identical to previous data) but was apparent by immunohistochemistry. Our explanation as to why H3 was not seen in the western blot assay of normal liver was that the normal liver nuclei might be removed intact during the total cell extraction procedure so that H3 was not exposed (11). We also identified H3 to be tightly bound to DNA so that it could not be recognized by anti-H3 antibodies in normal liver (8). In this study, through immunohistochemistry we detected H3 in normal liver tissues and further confirmed that expression of H3 was up-regulated in HCC.
The role of histone modification, especially acetylation, in the development of cancer and other diseases has been investigated. Aberrant acetylation or deacetylation of histone is implicated in cancer progression and other diseases (5). Histone deacetylation causes gene silencing and is thought to be the molecular pathway of HCC tumorigenesis (13). HDACs have important roles in histone modification and chromatin remodeling, and abnormal HDAC expression might cause aberrant acetylation or deacetylation of histone and play roles in tumor transformation. Some types of human cancer cells have been reported to have higher HDAC1 activity than normal cells (14, 15), while in human HCC, higher HDAC1 expression also indicates aggressiveness, cell de-differentiation, and poor prognosis (16). In this study, we showed that higher HDAC1 activity, as well as H3 hypoacetylation, might be a step towards the development of human HCC.
The mechanism of HDAC involvement in tumor transformation of human HCC is still under investigation, but might be related to aberrant cell-cycle regulation and failed apoptosis. It has been reported that up-regulation of HDAC8 promotes proliferation and inhibits apoptosis in HCC (17). The HDAC inhibitor trichostatin A induces G2/M phase arrest and apoptosis in YD-10B oral squamous carcinoma cells (18). Alterations of histone-modifier genes play an important role in gastric carcinogenesis, contributing to MYC (v-myc avian myelocytomatosis viral oncogene homolog) and CDKN1A deregulation (19). In HCC cells, up-regulation of HDAC1-3 reduces expression of microRNA-449. MicroRNA-449 binds c-MET mRNA to reduce its levels, promoting apoptosis and reducing proliferation of liver cells. Reduced expression of microRNA-449 induced strong effects on proliferation and survival in HCC (13). These studies suggest that overexpression of HDACs might promote tumor transformation by inducing cell proliferation, reducing apoptosis or increasing histone deacetylation. The same phenomenon might occur in human HCC, which we are currently investigating.
Recently, HDAC inhibitors have been used in experiments for cancer therapy. Examples include microRNA-34A regulation of therapy resistance by targeting HDAC1 and HDAC7 in breast cancer (20); a novel class I HDAC inhibitor, MPT0G030, induces cell apoptosis and differentiation in human colorectal cancer cells (21); combined mammalian target of rapamycin and HDAC inhibition has been investigated in prostate cancer therapy (22); belinostat has been used for the treatment of peripheral T-cell lymphomas (23); and targeting of histone deacetylases in brain tumors (24). We agree that HDAC inhibitors might be a good choice for cancer therapy, since suppression of HDAC might increase the acetylation level of H3, which in turn will activate cell-cycle arrest and apoptosis of cancer cells.
In conclusion, overexpression of HDAC1 in HCC might catalyze histone deacetylation and result in H3 hypoacetylation and H3 modulation. This study confirmed a previous finding that H3 modulation is related to overexpression of HDAC1 in human HCC and showed that by inducing H3 hypoacetylation, HDAC1 might play important roles in tumor transformation in human HCC.
Acknowledgements
The Authors greatly appreciate the skillful laboratory assistance of Miss You-Yin Chen.
- Received October 23, 2014.
- Revision received November 10, 2014.
- Accepted November 14, 2014.
- Copyright © 2015 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved