

Abstract
Background: The murine model of 4-nitroquinoline 1-oxide (4-NQO)-induced oral and esophageal cancer is frequently used to assess the effects of different cancer prevention/ therapy agents in vivo, but the molecular mechanisms in those 4-NQO-induced carcinogenesis are unknown. This study investigated aberrant expression of cell growth-critical genes in 4-NQO-induced oral and esophageal cancer tissues in mice compared to those present in the human disease for association with survival of patients. Materials and Methods: C57LB6/129Sv mice were given 4-NQO in their drinking water to induce oral and esophageal cancer. Quantitative-reverse transcription polymerase chain reaction (qRT-PCR), western blot, and immunohistochemistry were used to detect gene expression in the cancer tissues from mice and in 4-NQO-treated human esophageal cancer cell lines and esophageal cancer tissues. Methylation-specific PCR and DNA sequencing were performed to assess methylation of the Rarb2 promoter in murine tissues. Kaplan-Meier analysis was performed to associate gene expression in esophageal cancer tissues with survival data for patients with esophageal cancer. Results: 4-NQO dose-dependently induced pre-malignant and malignant lesions in the oral cavity and esophagus in mice that pathologically and morphologically mimicked human oral and esophageal cancer. Molecularly, 4-NQO inhibited Rarβ2 but induced expression of phosphorylated extracellular-signal-regulated kinase-1 and -2 (p-ERK1/2) and Cox2 proteins and Rarβ2 gene promoter methylation in murine tumors. In vitro treatment with 4-NQO altered expression of RARβ2, p-ERK1/2, and COX2 in human esophageal cancer cells. In tissues from 90 patients with esophageal cancer, expression of p-ERK1/2 and COX2 was up-regulated, and p-ERK1/2 expression was associated with advanced clinical tumor stage and consumption of hot beverages, while COX2 expression was associated with tumor de-differentiation in esophageal cancer. Furthermore, expression of p-ERK1/2 was associated with a worse overall survival rate of patients (p=0.014), whereas the association of COX2 expression with worse overall survival rate did not reach statistical significance (p=0.19). Knockdown of COX2 expression using transient transfection of a COX2 antisense expression vector inhibited Ki67 expression, an indicator of cell proliferation, in human esophageal cancer cells. Conclusion: 4-NQO-induced cancer in oral cavity and esophagus of mice not only pathologically and morphologically mimicked human oral and esophageal cancer, but also shared some molecular alterations (e.g. aberrant expression of Rarb2, p-ERK1/2, and Cox2). This study further demonstrated that targeting of the altered RARβ2-led gene pathway could effectively suppress the development of this deadly type of cancer.
- 4-NQO
- animal model
- oral cancer
- esophageal cancer RARβ2
- and p-ERK1/2
- COX2
Oral and esophageal carcinomas are significant health problems that account for approximately 900,000 new cancer cases annually worldwide (1, 2). Oral cancer usually occurs as squamous cell carcinoma (SCC), whereas esophageal carcinoma occurs as either SCC or adenocarcinoma (1-7). Esophageal SCC accounts for approximately one third of esophageal cancer cases in the United States but represents more than 90% cases of esophageal cancer worldwide (6, 7). The greatest risk factors for oral and esophageal SCCs are tobacco smoke and heavy alcohol consumption, which together account for almost 90% of all cases; a diet lacking fresh fruits and vegetables also increases the risk for these types of cancer (6, 7). Moreover, studies have demonstrated that vitamin A deficiency is also closely related to the development and progression of oral and esophageal SCCs in certain populations (8, 9). The 5-year survival rate associated with head and neck cancer is about 50%, but that for patients with esophageal SCC is poor (only 5%-15% at five years) (1-8). To effectively reduce the mortality rate in these patients, we need to better understand the molecular mechanisms underlying the development of these deadly diseases and translate that knowledge into novel approaches for their early diagnosis, treatment, and prevention. Toward this end, the murine model of 4-nitroquinoline 1-oxide (4-NQO)-induced oral and esophageal cancer is frequently used to assess the effects of different cancer preventive and therapy agents in vivo, especially for oral cancer (10-20). 4-NQO is quinoline derivative and a tumorigenic compound commonly used in such assessments in animal models. However, the molecular mechanisms responsible for 4-NQO-induced oral and esophageal cancer remain to be defined.
In the development of human oral and esophageal SCCs, multiple gene alterations and genetic modifications occur (6, 7). For example, our group and others have demonstrated that chemical carcinogens present in tobacco and environmental pollutant [e.g. benzo(a)pyrene diolepoxide (BPDE)] inhibited retinoic acid receptor β2 (RARβ2) but induced expression of phosphorylated extracellular-signal-regulated kinase-1 and -2 (p-ERK1/2) and cyclooxygenase-2 (COX2) in oral and esophageal cancer cell lines (21-24) and that loss of RARβ2 expression is a common and early event in different human cancer tissues, including oral and esophageal SCC (25-27). BPDE treatment methylated the RARβ2 gene promoter according to one study (28), while other studies also showed that 4-NQO was able to inhibit Rarβ2 but induce Cox2 expression in C57BL6 mice (11). Thus, in the current study, we explored the molecular events responsible for the activity of 4-NQO in vitro and in vivo by analyzing the expression of the above-named genes in 4-NQO-induced murine tumor tissues and human esophageal cancer cell lines and tissue specimens and then by associating the expression of these genes with clinicopathological and survival data from patients with esophageal cancer.
Materials and Methods
Animal experiments. Six-week-old C57LB6/129Sv mice were housed in plastic cages in an air-conditioned room with a 12 h light–dark cycle and a basal diet (Taklad Global 19% Protein Extruded Rodent Diet 2919; diet and mice both from Harlan Laboratories, Houston, TX, USA); sterilized water was available ad libitum. The animal experimental protocol was approved by our Institutional Animal Care and Use Committee (#10-10-09331). The mice were randomly divided into four groups; three groups each received drinking water containing different concentrations (15, 30, and 45 μg/ml) of 4-NQO and the fourth group received no 4-NQO (control). The stock solution for carcinogen 4-NQO was prepared in propylene glycol (both from Sigma, St. Louis, MO, USA) at 6 mg/ml and diluted in the drinking water once a week. All groups received the same volume of propylene glycol in the solution. Consumption was recorded to estimate the intake of 4-NQO. The mice were allowed access to the drinking water at all the times. After eight weeks of 4-NQO treatment, the mice were followed-up for another 16 weeks. Body weight of the mice was measured at the beginning and the end of the experiment.
Two hours before the experiments ended, the mice were injected with 1 mg/100 g body weight bromodeoxyuridine (BrdU; Invitrogen, Carlsbad, CA, USA) via the tail vein and then sacrificed by CO2 inhalation. The oral cavity, esophagus, and stomach were inspected for formation of pre-malignant and malignant lesions and then quickly dissected, fixed in freshly-made 4% paraformaldehyde solution, and routinely processed for embedding in paraffin.
Quantitative-reverse transcription polymerase chain reaction (qRT-PCR) analysis of RARβ2 and COX2 mRNA expression. qRT-PCR was performed to assess expression of RARβ2 and COX2 mRNA in oral and esophageal cancer tissues of the experimental mice and in human esophageal cancer cell lines. Total RNA was isolated from the formalin-fixed and paraffin-embedded tissue sections using a MasterPure complete DNA and RNA purification kit (Epicentre Biotechnologies, Epicentre, WI, USA) or using TRIzol Reagent (Invitrogen, Grand Island, NY, USA) for cell lines. ABI 7300 Real-Time PCR system and Brilliant II SYBR Green QPCR Master Mix kit (Agilent Technologies, CA, USA) were used for qPCR according to a standard method. The data were presented as CT number for each sample. The primers used for mRarβ2 were 5’-CAGTGGATTCACCCAGGCCG-3’ and 5’-GGACGAGCTCCTCAGAGCTGG-3’, the primers used for mCox2 were 5’-ACGCTTCTCCCTGAA GCCGTAC-3’ and 5’-GTAGAGGGCTTTCAATTCTGCAGCC-3’, and the primers used for m36B gene were 5’-ACCGCCTGGTTCTCCTATAAAAGGC-3’ and 5’-GCGGTGCGTCAGGGATTGCC-3’. The primers used for human RARβ2 were according to our previous study (21, 28).
Methylation-specific PCR (MSP), PCR cloning, and DNA sequencing. Genomic DNA from the formalin-fixed and paraffin embedded mouse tissues was extracted and then subjected to methylation-specific PCR analysis with an MSP kit (Zymed, South San Francisco, CA, USA) according to our previous study (28). The primers used to amplify the methylated mRarβ2 gene were 5’-TCGTGGTTTTTTTGTGCGGTTC-3’ and 5’-CAACATACAAAAAAAAAAAC TCGCG-3’. The primers used to amplify the unmethylated mRarβ2 gene were 5’-TT GTGGATTTTTTTGTGTGGTTTG-3’ and 5’-CAACATACAAAAAAAAAAACTCACAA-3’. GeneAmp 9700 PCR system (Applied Biosystems) was used and the PCR products were run on 2% agarose gel and visualized under UV illumination.
The PCR products from MSP analysis were then purified and cloned into pCR™2.1-TOPO® vector (Invitrogen). The vectors were then transferred into Escherichia coli and amplified for plasmid DNA preparation using a PureLink Quick Plasmid Miniprep Kit (Invitrogen). These plasmid DNA samples were then sequenced using M13 primer in our DNA sequencing facility at the MD Anderson Cancer Center.
Cell lines and culture. Human esophageal squamous cell cancer cell lines TE-3, TE-8, and HCE-4 and the esophageal adenocarcinoma cell line SKGT-4 were used in our previous studies (21-24) and cultivated in tissue culture dishes with Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere of 95% air and 5% CO2. An SV-40-immortalized human esophageal epithelium HET-1A cell line (21) was grown in keratinocyte serum-free medium (Invitrogen) at 37°C in a humidified atmosphere of 95% air and 5% CO2. For 4-NQO treatments, these cell lines were seeded overnight and then treated with 4-NQO (Sigma). In the dose-dependent model, cell lines were treated with a concentration of 4-NQO between 0.01 μM and 2 μM for 24 h, control cell lines received no 4-NQO but propylene glycol amounts equal to those of the treated cell lines. In the time-dependent model, HET-1A cells were treated with 0.25 μM 4-NQO, but TE-3 and TE-8 cells were treated with 0.5 μM 4-NQO for up to 48 h, which is the optimal dose for these cell lines according to the dose-dependent study.
Protein extraction and western blot analysis. Total cellular and nuclear proteins were isolated as described previously (21-24). The protein concentration was then measured with a BioRad Protein Assay Kit II (BioRad Laboratories, Hercules, CA, USA) according to the manufacturer's recommendations. Samples containing 50 μg of protein from the control or treated cells were separated in 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis gels and then transferred electrophoretically onto Hybond-C nitrocellulose membranes (GE Healthcare, Arlington Heights, IL, USA) at 500 mA for 2 h at 4°C. The membranes were subsequently stained with 0.5% ponceau S solution containing 1% acetic acid to confirm that proteins were loaded equally and to verify transfer efficiency. The membranes were next incubated overnight in a blocking solution containing 5% bovine skim milk and 0.1% Tween 20 in PBS at 4°C. The next day, the membranes were incubated with a primary antibody for 2 h at room temperature. The antibodies used were anti-c-FOS (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA); anti-COX2 (BD Transduction Laboratories, Lexington, KY, USA); anti-ERK1/2, and p-ERK1/2 (Cell Signaling Technology, Beverly, MA, USA); and anti-β-actin (Sigma). The membranes were washed in PBS and incubated for 1.5 h with a horse anti-mouse or goat anti-rabbit secondary antibody (GE Healthcare) diluted 1:5000. The membranes were then incubated with enhanced chemiluminescence solution (GE Healthcare) for 1-2 min and exposed to an X-ray film.
Antisense Cox2 cDNA transfection and immunocytochemical staining. Human esophageal cancer cells from lines TE-8, SKGT-4, and HCE-4 were grown in monolayer overnight and transiently transfected with either mammalian expression vector pCMS/EGFP (BD Clontech, San Diego, CA) plus pcDNA3.1 vector (Invitrogen) or pcDNA3.1/COX2AS, a Cox2 antisense cDNA vector [see our previous study (23, 24)] using Lipofectamine 2000. Forty-eight hours later, the cells were fixed with 4% paraformaldehyde and permeabilized in 0.5% Triton X-100 for 10 min each at room temperature. The fixed cells were then subjected to Ki-67 immunostaining, as described previously (24). The Ki-67 antibody was obtained from Vector Laboratories (Burlingame, CA, USA) and used at a dilution of 1:50 in PBS. Subsequently, more than 200 cells in 10 fields were counted (under a ×20 objective lens) for positive staining for green fluorescent protein (GFP) as well as for positive or negative Ki-67 (red) staining in these cells. The percentage of cell proliferation relative to that of the control vector was calculated from the equation: % proliferation=NT/NV×100, where NV and NT were the numbers of Ki-67-positive and GPF-positive cells of control vector and pcDNA3.1/COX2AS-transfected cells, respectively.
Human esophageal cancer tissue specimens. A protocol for the use of patients' tissue samples in this study was approved by our Institutional Review Board (#LAB-96-175). We obtained paraffin-embedded tissue samples from 90 consecutive patients with esophageal SCC who had undergone surgery without preoperative chemotherapy or radiotherapy between 2003 and 2005 at The First Affiliated Hospital of Anhui Medical University, Hefei, China. Corresponding specimens from each patient's distant (≥4 cm from the tumor lesions) normal tissue were also collected in each case. The patients' ages ranged from 41 to 76 years (median=59 years). Clinicopathological data also were collected from these patients' medical records, and the patients were followed-up until August 2010.
Preparation of human esophageal cancer and normal tissue microarrays. The formalin-fixed and paraffin-embedded esophageal cancer and normal tissue specimens were sectioned and stained with hematoxylin and eosin (HE). The most representative areas of the HE-stained sections were labeled with a localizer under a microscope. Three cores per esophageal carcinoma paraffin block and one core per normal mucosa paraffin block were used to construct the tissue microarray with a Liaoning Hengtai HT-1 tissue arrayer (Liaoning, China).
Immunohistochemistry. A paraffin block of each tissue microarray was cut into 4-μm thick sections, and every tenth section was then stained with HE for reference. These tissue microarray sections were immunostained for p-ERK1/2 and COX2 expression using the ABC staining technique. A rabbit polyclonal antibody to p-ERK1/2 (Cat #9101s) was purchased from Cell Signaling Technology and used at a dilution of 1:20, and a mouse monoclonal antibody against COX2 (clone #SP21) was purchased from BD Transduction Laboratories and used at a dilution of 1:50. The ABC kit was purchased from Vector Laboratories, and 3-amino-9-ethylcarbazole (AEC) staining reagent was purchased from Sigma.
Review and scoring of immunostained sections. The immunostained sections were reviewed and scored in a blinded fashion semi-quantitatively, independently by two investigators (YZ and XX) through review of both intensity and the percentage of positive cells in each tissue core. More than five microscopic fields were observed for each core at ×400 magnification. For staining intensity, 0 was denoted as no staining, 1 was weakly positive staining with faint red, 2 was moderately positive, and 3 was strongly positive with dark red staining. The percentage of positive cells was counted for 100 tumor cells per field, five fields in each tissue core. The number of positive cells was visually evaluated as follows: <10% positive cells as 0 (negative), <25% positive cells as 1 (weak), <50% positive cells as 2 (moderate), and ≥50% positive cells as 3 (strong). The sum (staining index) of staining intensity and positive cell scores was used to record the final result for each section: ≥3 as high expression and <3 as low expression.
Statistics analysis. Differences between two groups were compared using Pearson's chi-square test. Overall survival curves were plotted separately for patients with low versus high expression of each gene using the Kaplan–Meier method. The log-rank test was used to compare the survival times between the patients with high and those with low expression of specific genes. Overall survival duration was calculated from surgery to the date of death or end of follow-up. All statistical analyses were performed using the SPSS statistical package, version 13 (SPSS Inc., Chicago, IL, USA). A p-value <0.05 was considered statistically significant.

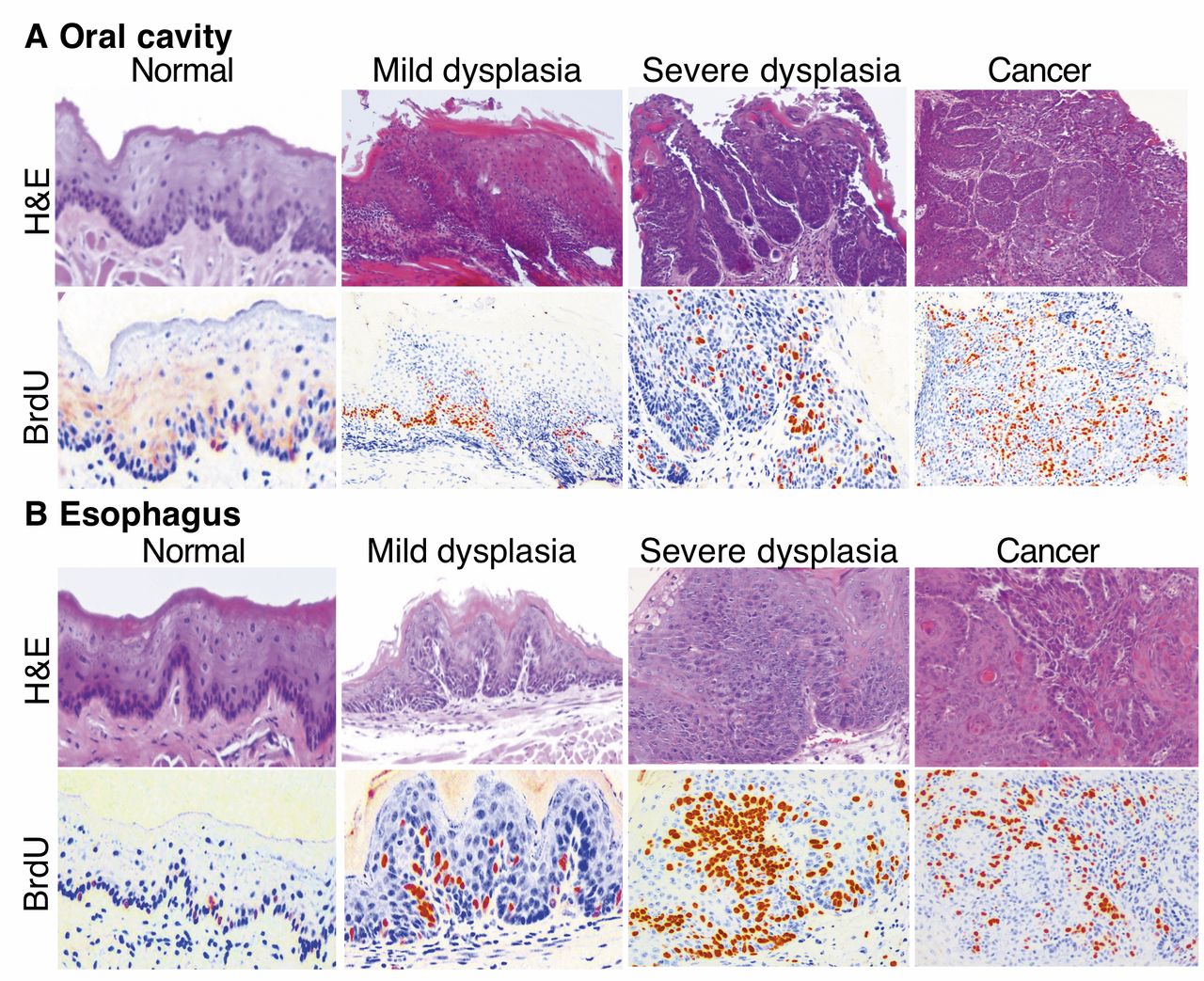
A murine model of 4-nitroquinoline 1-oxide (4-NQO)-induced oral and esophageal squamous cell carcinoma (SCC). A total of 40 mice were given drinking water containing or not-containing 4-NQO (15 μg/ml, 30 μg/ml, or 45 μg/ml) for eight weeks, followed by regular drinking water and observation for another 16 weeks. Two hours before mice were sacrificed, Bromodeoxyuridine (BrdU) was injected into their tail vein. After the mice were sacrificed, lesions from the oral cavity and esophagus were inspected and resected and then fixed in 10% formalin and embedded in paraffin for sectioning and (HE) and BrdU staining. Note: Reddish and yellowish color indicates BrdU-positive staining, and blue color is HE-counterstaining of the nuclei of the cells.
Data on 4-nitroquinoline 1-oxide (4-NQO)-induced oral and esophageal cancer development in mice.
Results
4-NQO induction of oral and esophageal tumorigenesis in mice. 4-NQO-induced oral and esophageal cancer in mice is an established animal model for study of human diseases. In this study, we induced tumors in this manner. Our data showed that 4-NQO dose-dependently induced pre-malignant and malignant lesions in murine oral cavity and esophagus after 8-week treatment with 4-NQO administered in the animals' drinking water followed by a16-week waiting period. Table I summarizes the body weight changes of the mice before and after the experiments; results indicated that body weight of 4-NQO-treated mice showed less gain than that of control mice in a dose-dependent manner. Table I summarizes the tumor induction rates in the 4-NQO-treated mice. HE staining data revealed that the 4-NQO-induced oral and esophageal lesions mimicked human oral and esophageal cancer morphologically, e.g. 4-NQO induced mild and severe dysplasia and SCC in the oral cavity and esophagus (Figure 1). BrdU immunostaining showed that cells in 4-NQO-induced lesions had greater BrdU incorporation into DNA than did normal tissues.
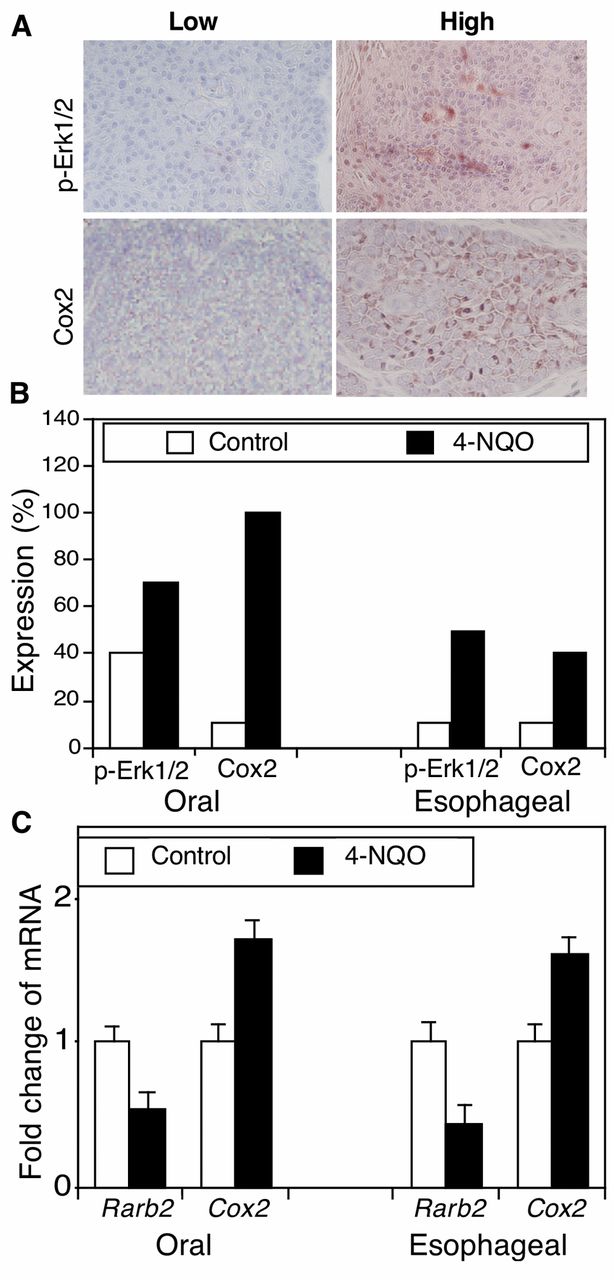
Aberrant expression of cell growth-critical genes in 4-NQO-induced oral and esophageal cancer in mice. To better-understand the underlying molecular mechanism for 4-NQO-induced carcinogenesis, we detected expression of p-Erk1/2 and Cox2 protein and Rarβ2 mRNA in these mouse oral and esophageal carcinomas. The immunostained sections were reviewed and scored as showing high or low protein expression (Figure 2A and B). In particular, p-Erk1/2 protein was weakly-expressed in 4 out of 10 oral tissues in the control mice but highly expressed in 7 out of 10 of oral SCCs treated with 45 μg/ml 4-NQO. Similarly, p-Erk1/2 protein was only weakly-expressed in 1 out of 10 esophageal epithelium tissue samples in the control mice but highly expressed in 5 out of 10 esophageal SCCs from mice treated with 45 μg/ml 4-NQO (Figure 2B). Furthermore, the Cox2 protein was expressed in 1 out 10 of oral tissues in the control mice but in all 10 oral SCCs after 4-NQO treatment. In the esophagus, 1 out of 10 normal tissue samples expressed Cox2, but all 10 tumor tissues expressed Cox2 (Figure 2B). In contrast, compared with expression in tissues from control mice, Rarβ2 mRNA expression was reduced in both oral and esophageal tumors (Figure 2C).
4-NQO-induced methylation of Rarβ2 gene promoter in murine tumors. In our previous study, we showed that the tobacco carcinogen BPDE inhibited RARβ2 expression through methylation of RARβ2 gene promoter (28) and then, in turn, up-regulated expression of p-ERK1/2 and COX2 proteins in human esophageal cancer cell lines (21-24). In the current study, we also found that 4-NQO suppressed Rarβ2 expression in these murine oral and esophageal tumors. Thus, we performed methylation-specific PCR and DNA sequence analyses to detect RARβ2 gene promoter methylation in these mouse tissues. Our data showed that 4-NQO did indeed induce Rarβ2 gene promoter methylation in five randomly selected cases each of oral and esophageal SCC tissues but not in the five control normal tissues. DNA sequence data showed that a representative clone of PCR products had methylation at five nucleotide sites but we did not find methylation in normal tissues. Figure 3 shows only data on esophageal tissues.
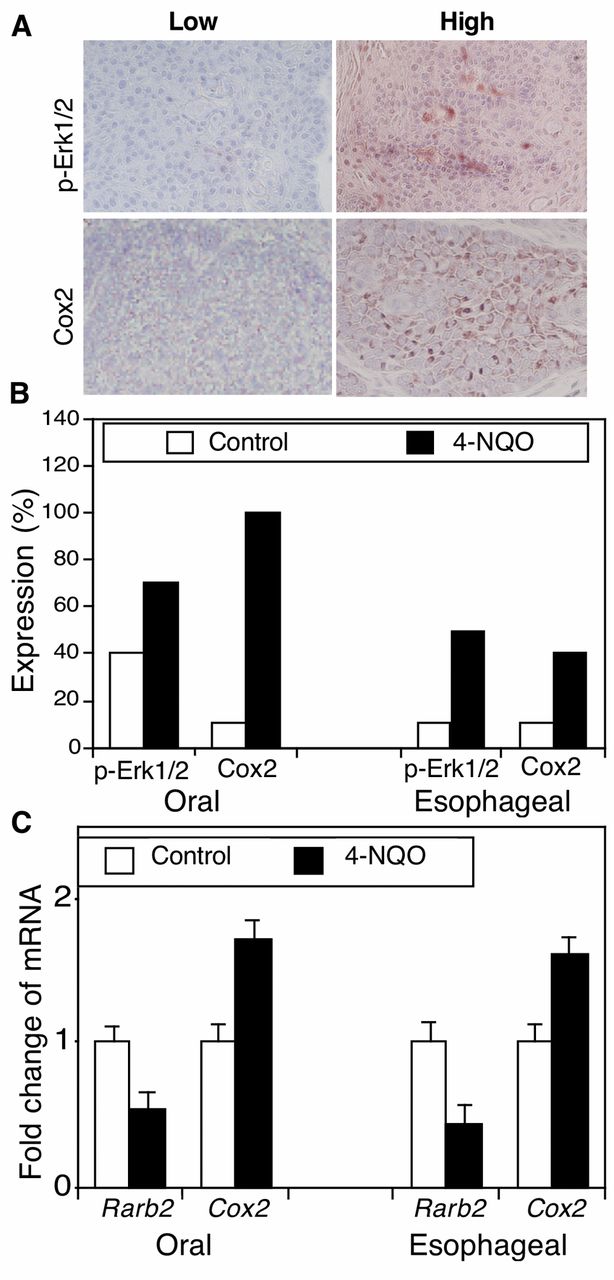
4-nitroquinoline 1-oxide (4-NQO)-altered expression of phosphorylated extracellular-signal-regulated kinase-1 and -2 (p-Erk1/2) and cyclooxygenase-2 (Cox2) in murine tumor tissues. C57BL6 mice were treated with propylene glycol (vehicle) or 4-NQO (45 μg/ml) in drinking water for eight weeks and then received regular drinking water for an additional 16 weeks. A: Immunohistochemistry. Tissue sections from these mice were immunohistochemically stained with antibodies against p-Erk1/2 or COX2 and were then reviewed and scored for expression of each protein. Representative photographs were taken and are presented ×200. B: Semi-quantitative data from immunohistochemistry. *p<0.05 Compared with control tissues using Fisher's exact test. C: Quantitative reverse transcription-polymerase chain reaction (qRT-PCR). Formalin-fixed and paraffin-embedded tissue sections were subjected to RNA isolation and qRT-PCR analysis. *p<0.05 Compared with the control tissues using Student's t-test.

4-nitroquinoline 1-oxide (4-NQO)-induced methylation of Rarβ2 gene promoter in murine tumors. Mice were treated with 45 μg/ml of 4-NQO in the drinking water for eight weeks and then received regular drinking water for another 16 weeks. The mice were then sacrificed, and five cases of normal and cancerous tissues were used for this analysis. A: Methylation-specific PCR (MSP). Murine tissue specimens were subjected to DNA extraction, and genomic DNA was then subjected to bisulfate treatment using a methylation kit from Zymed and PCR analysis of Rarβ2 gene promoter methylation. Note: M, Methylated DNA primers; U, Un-methylated DNA primers. B: DNA sequencing. The MSP products from A were cloned, amplified, and then sequenced in our Institutional DNA sequencing facility. Compared with normal tissues (not treated with 4-NQO), tissues from the 4-NQO-treated mice showed methylation (arrows). Note: Bisulfite converted all C to T, but methylated C cannot be converted, which is the principle of the MSP assay. The graphs show the antisense sequences due to the DNA cloning process.
4-NQO altered gene expression in human esophageal cancer cells. Next, we assessed the effects of 4-NQO on regulation of gene expression in human esophageal cancer cell lines. Our data showed that 4-NQO inhibited expression of RARβ2 mRNA but induced expression of COX2 mRNA in SV40-immortalized esophageal cells (HET-1A) and esophageal SCC TE-3 and TE-8 cell lines (Figure 4A and B). However, the tolerance (toxicity) of each of the cell lines to 4-NQO treatment differed, i.e. HET-1A cells were more sensitive to 4-NQO treatment than were TE-3 and TE-8 cells (data not shown). Moreover, we also determined whether the RARβ2-led gene pathway was affected by 4-NQO treatment and found that indeed, 4-NQO treatment inhibited RARβ2 expression and up-regulated p-ERK1/2, c-FOS and COX2 expression in RARβ2-positive HET-1A and TE-3 cells, but such effects were much less in RARβ2 negative TE-8 cells (Figure 4C), indicating that expression of RAR-β2 plays an important role in 4-NQO-induced COX2 expression.
Aberrant expression of p-ERK1/2 and COX2 in human esophageal cancer tissues and association with clinicopathological data and survival of patients with esophageal cancer. We also detected expression of p-ERK1/2 and COX2 in tissues from 90 patients with esophageal SCC; the data showed that p-ERK1/2 expression in esophageal SCC tissue was higher (18.9%, 17/90) than in the distant normal esophageal mucosa (5.8%, 4/69; p=0.018). Expression of the COX2 protein in esophageal SCC tissue was also higher (42.2%, 38/90) than in normal esophageal mucosa (20.3%, 14/69; p=0.004) (Figure 5). Next, we determined the association of altered expression of p-ERK1/2 and COX2 proteins with clinicopathological parameters of patients with esophageal cancer. Table II shows that expression of p-ERK1/2 in esophageal SCC tissues was associated with advanced clinical stage and with consumption of hot beverages, while COX2 expression was associated with tumor de-differentiation. However, expression of neither of these proteins was associated with patients' tobacco smoking and alcohol consumption habits, which may suggest that risk factors for esophageal cancer in this Chinese population differ from these in Western populations.

4-nitroquinoline 1-oxide (4-NQO)-altered gene expressions in human esophageal cell lines. A: Quantitative reverse transcription-polymerase chain reaction (qRT-PCR). An SV-40-immortalized human esophageal epithelial cell line HET-1A and esophageal squamous cell carcinoma TE-3 and TE-8 cell lines were grown and treated with different concentrations of 4-NQO for 24 h. Total cellular RNA was then isolated from these cells and subjected to qRT-PCR analyses. The data were normalized to those for a housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and then summarized as fold changes compared with the control experiments. B: qRT-PCR. These cells were grown and treated with 0.5 μM 4-NQO for different periods of time, and RNA was then isolated from these cells for qRT-PCR analysis. C: Western blot. These three cell lines were treated with different concentration of 4-NQO (from 0.01 μM to 2 μM) for 24 h, and cells were cultured with equal volumes of propylene glycol in DMEM as a control. At the end of the experiments, total cellular protein was extracted and subjected to western blot.

Differential expression of phosphorylated extracellular-signal-regulated kinase-1 and -2 (p-ERK1/2) and cyclooxygenase-2 (COX2) proteins in normal and cancerous human esophageal tissue specimens. Tissue microarray sections from patients with esophageal cancer were immunohistochemically stained with antibodies against p-ERK1/2 and COX2 and then reviewed and scored for expression of each protein. Representative photographs were taken and are shown ×200.
We also collected prognostic data from these patients for association with expression of p-ERK1/2 and COX2. Our data show that the survival rate of these patients was 88.9% (80/90) at one year and 63.3% (57/90) at three years. In contrast, 51.1% (46/90) of the patients were dead at the last following-up. Expression of p-ERK1/2 was significantly associated with poor overall survival of patients with esophageal cancer (p=0.014), but the association of COX2 expression with poor overall survival did not reach statistical significance (p=0.19; Figure 6).
Knockdown of COX2 expression in the regulation of esophageal cancer cell growth. We performed an additional experiment to assess whether knockdown of COX2 expression is able to inhibit growth of esophageal cancer cells using transient transfection of a COX2 antisense expression vector (23, 24). Our data show that knockdown of COX2 expression suppressed Ki67 expression (an indicator of cell proliferation) (Figure 7). However, in COX2-weakly expressing SKGT-4 cells, antisense COX2 cDNA transfection had less effect on inhibition of tumor cell growth (Figure 7).
Discussion
In the current study, we demonstrated that 4-NQO-induced pre-malignant and malignant lesions in oral cavity and esophagus in mice not only pathologically and morphologically mimicked human oral and esophageal cancers but also shared some molecular alterations (e.g. altered expression of Rarβ2, p-Erk1/2, and Cox2) with human cancer. Our results further suggest that targeting of the altered RARβ2-led gene pathway may effectively suppress development of oral and esophageal cancer in humans.
4-NQO is a synthetic carcinogen first investigated by Nakahara et al. (29) and shown to induce oral cancer by Ohne et al. (30). To date, numerous studies on 4-NQO induction of oral and esophageal carcinoma in rat and mouse models have been reported. For example, Hasina et al. demonstrated that after eight weeks of 100 μg/ml 4-NQO in their drinking water followed by 24 weeks of follow-up, experimental CBA mice exhibited a hyperkeratosis, dysplasia, and head and neck SCC (14). Shiotani et al. demonstrated that tongue SCC lesions developed in Fischer 344 rats after 4-NQO treatment (31). Tang et al. showed that 4-NQO treatment induced more multifocal pre-cancerous and cancer lesions in the tongue and esophagus of the transgenic mice (11). Yamamoto et al. reported that tongue SCCs were induced in rats after 4-NQO treatment (18, 19) and also found expression of prostaglandin-E2 and prostaglandin-E2 receptor in tumors after 4-NQO treatment. Furthermore, several different studies used this animal model to assess the effects of different chemopreventive agents (10-20, 31, 32). However, the molecular mechanisms responsible for 4-NQO-induced oral and esophageal cancer have yet to be defined. Thus, we took this opportunity to compare the gene expressions in 4-NOQ-induced murine esophageal cancer with this type of cancer in humans.

Association of phosphorylated extracellular-signal-regulated kinase-1 and -2 (p-ERK1/2) and cyclooxygenase-2 (COX2) expression with overall survival of patients with esophageal cancer. Overall survival of the patients was calculated from the date of surgery to the date of death. Kaplan–Meier curves were plotted and the log-rank test was performed. A: p-ERK1/2. B: COX-2.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of cyclooxygenase-2 (COX2) gene knockdown on regulation of esophageal cancer cell growth. Esophageal squamous cell carcinoma (SCC) TE-8 and HCE-4 cells and esophageal adenocarcinoma SKGT-4 cells were grown and transiently transfected with antisense COX2 cDNA plus pCMS/EGFP vector for 48 h, and the cells were subjected to immunostaining for Ki67 expression. Approximately 200 green fluorescent protein-positive cells from these transfections were identified and assessed for Ki-67 expression (red). Data are summarized as means±SD and calculated as a percentage of the control vector-transfected cells. *p<0.05 compared with control cells.
In our study, we found that expression of p-ERK1/2 and COX2 proteins was up-regulated in 4-NQO-induced oral and esophageal SCC tissues, which confirmed findings of previous studies that Egfr and Cox2 were overexpressed in 4-NQO-induced oral SCC tissues in C57BL6 mice (10, 11) and in rats (18, 19). Moreover, Yoshida et al. demonstrated that a Cox2 inhibitor inhibited 4-NQO-induced rat tongue carcinogenesis by suppressing cell proliferation activity and Cox2 and cytokine-inducible nitric oxide synthase (iNos) expression (32), whereas Shiotani et al. showed that Cox2 expression was barely detected in the normal tongue epithelium but increased 6-fold in tumor tissues (31). However, our current study revealed that loss of Rarβ2 expression after in vivo 4-NQO treatment was due to methylation of the Rarβ2 gene promoter, which further confirmed our previous in vitro data that the tobacco carcinogen BPDE induced methylation of the Rarβ2 gene promoter by recruiting DNA (cytosine-5)-methyltransferase 3A (DNMT3a) in esophageal cancer cell lines (28).
We also linked the loss of RARβ2 expression to increased p-ERK1/2 and COX2 expressions in esophageal cell lines in vitro. Indeed, a previous study showed that BPDE can induce methylation of the Rarβ2 gene promoter in mouse lung tissues (33), and our data (28) showed that BPDE-induced COX2 expression was mediated by inhibition of RARβ2 and consequently, by induction of EGFR, phosphorylated ERK1/2, and activator protein-1 (AP-1) expression in esophageal SCC cell lines (21-24). Esophageal cancer cells that do not express RARβ2 did not respond to BPDE treatment by inducing COX2 expression (24). Tang et al. also showed higher Cox2 expression but lower Rarβ2 expression in tongue tissues of 4-NQO-treated mice than in tissues from control mice (11). Our previous study also demonstrated that RARβ2 expression was inversely associated with COX2 expression in oral pre-malignant lesions and that retinoic acid treatment induced RARβ2 expression but down-regulated COX2 expression in patients (24). Taken together, these findings suggest that the altered RARβ2-led gene pathway plays a role in development of oral and esophageal cancer.
Association of phosphorylated extracellular-signal-regulated kinase 1 and 2 (p-ERK1/2) and cyclooxygenase 2 (COX2) expression with clinicopathological data for patients with esophageal cancer.
Furthermore, our ex vivo data demonstrated that expression of p-ERK1/2 and COX2 was upregulated in dysplastic and malignant esophageal squamous cell epithelia compared with expression in normal mucosa. Our previous studies and those of others demonstrated the loss of RARβ2 expression in oral and esophageal SCC tissues (25-27). ERK1/2 is a member of the mitogen-activated protein kinases superfamily. ERK1/2 protein is activated through phosphorylation to induce cell growth-related genes such as COX2 and cyclin-D1 in development of various types of human cancers (34). Previous studies showed that p-ERK1/2 is expressed in oral and esophageal SCC (35-39). Furthermore, COX2 is the rate-limiting enzyme in arachidonic acid metabolism and is inducible by various agents (such as growth factors and tumor promoters) (35). Overexpression of COX2 was reported in oral and esophageal cancer and promoted cell growth, invasion, and metastasis of these types of cancers (40-47).
Our recent study also showed that both p-ERK1/2 and COX2 were overexpressed in esophageal cancer tissues from American patients (39); in that study, expression of these two proteins was much higher than that found in the Chinese patients in the current study. The reason for the difference in protein expression is unclear but different risk factors between Chinese and American populations may play a role in esophageal cancer development (6). For example, our current data showed that expression of p-ERK1/2 and COX2 was not associated with tobacco smoking and alcohol consumption among the Chinese patients but that COX2 expression was associated with tobacco use in American and European patients with esophageal cancer (6,7), suggesting that this Chinese population had additional risk factors for esophageal cancer. Further study is needed to address this question by using a larger sample size.
In conclusion, our current study demonstrates that 4-NQO-induced pre-malignant and malignant lesions in oral cavity and esophagus in mice not only pathologically and morphologically mimicked human oral and esophageal cancer but also shared similar molecular alterations (e.g. altered Rarβ2, p-Erk1/2, and Cox2 expressions). However, this study was simply a proof-of-principle investigation because we did not perform a time course analysis in this animal experiment to assess spatiotemporal changes in expression of these genes. We also did not manipulate expression of these genes in 4-NQO-induced oral and esophageal cancer development in mice to assess whether targeting of these genes could prevent tumorigenesis in mice. Further studies are warranted to clarify these issues and to make this animal model more useful for research.
Acknowledgements
This work was supported in part by National Cancer Institute grant R01 CA117895, a Cancer Center Supporting Grant (CA16672) for our core DNA facility, and a seed grant from the Duncan Family Institute for Cancer Prevention and Risk Assessment. We thank the Department of Scientific Publications at The University of Texas MD Anderson Cancer Center for editing the manuscript.
Footnotes
-
Competing Interests
The Authors declare that they have no competing interests.
- Received March 21, 2013.
- Revision received April 15, 2013.
- Accepted April 17, 2013.
- Copyright © 2013 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved