

Abstract
Background/Aim: Causative agents most frequently encountered in systemic infections are bacteria, although fungi that cause invasive infections have also emerged, mostly in immune-compromised patients. The early detection and adequate treatment of bloodstream infections are critical for successful treatment. The aim of this study was to develop a rapid and efficient method for the detection and differentiation of the most common fungal pathogens. Materials and Methods: Real-time Polymerase chain reaction (PCR) and consecutive high-resolution melting analysis was used for the detection and differentiation of fungal pathogens. Results: The developed analysis procedure proved appropriate for discrimination of the ten most relevant Candida species, four Aspergillus species, and Cryptococcus neoformans. The sensitivity of the PCR reaction was 5, which is suitable for the detection of these fungi in blood. Conclusion: This technique is not adaptable as a general identification method, but it is highly useful when certain fungal species are to be expected in clinical samples.
- Candida
- Aspergillus
- ITS
- high-resolution melting (HRM)
- real-time PCR
Candida species are important fungal pathogens, particularly when they are involved in bloodstream infections (1). Such septic infections are associated with high rates of morbidity and mortality, especially when the host responses are weak, as in the case of immune-compromised individuals or patients with malignant conditions (2). Candida albicans is currently the most frequent causative agent of fungal sepsis, but the frequencies of invasive mycoses caused by C. parapsilosis, C. tropicalis, C. glabrata, C. kefyr, C. inconspicua and C. krusei have been reported to be increasing in several groups of patients (3). In vitro studies have indicated that emerging Candida species such as C. glabrata and C. krusei are demonstrating an increased incidence of resistance to anti-fungal agents, in particular to fluconazole (4, 5). The availability of fast and reliable procedures for the differentiation of Candida species has therefore become important for clinical laboratories. The diagnosis of candidaemia or haematogenous candidiasis can be problematic because of the low sensitivity of analysis via blood cultures (6). Non-culturing methods, including galactomannan antigenaemia, western blotting and polymerase chain reaction (PCR)-based approaches are therefore being developed and evaluated for the detection of mycotic infections (7). While RNA is very rarely used as a target in fungal identifications (8), the DNA coding sequences (9) and the internal-transcribed spacer (ITS) region in the ribosomal DNA (rDNA) appear to be excellent targets for PCR amplification (10). The ITS sequences are additionally used as reference standards for the validation of some new methods, e.g. repetitive-sequence PCR with electrospray ionization mass spectrometry (11).
The evaluation of the PCR product was initially based on electrophoresis with differential analysis of the amplicon length (12) or restriction fragment length polymorphism (13). Sequence analysis (14) and enzyme-linked immunosorbent assays (15) were later introduced and analysis is also possible with a combination of these methods (16). However, all of these investigations are time-consuming and labour-intensive relative to the real-time PCR method. Some of the assays involve the use of species-specific hybridization probes and differ in the technique applied, while the target and even the probe sequences are the same, e.g. TaqMan probes with the Perkin-Elmer PCR (17), and florescence resonance energy transfer (FRET) probes (18) and TaqMan probes (19) with the Roche LightCycler. In one of the most sophisticated approaches, two FRET probes were used and the differentiation was carried out via the melting analysis of the probes (20). The ITS targets sequences unique to fungal species, and differentiation is therefore possible by means of melting analysis of the whole amplicon produced by panfungal primers. This method has been described for the differentiation of dermatophyte fungi (21) and Candida species (22, 23). Species-specific primer pairs located in the ITS region have also been tested with melting analysis (24), by methods utilizing SybrGreen dye and the Roche LightCycler real-time PCR. New dyes (e.g. EvaGreen) and techniques (high-resolution melting analysis, HRM) have also recently been applied in diagnostic laboratories.
The aim of our study was to develop a rapid, sensitive and specific method for the detection of the most important Candida and Aspergillus species causing bloodstream infections. This was achieved via amplification of the ITS2 region and consecutive HRM analysis with use of the EvaGreen dye.
Materials and Methods
Fungal strains. Reference strains: Candida albicans ATCC 10231 and ATCC 14053, C. tropicalis ATCC 750, C. parapsilosis ATCC 22019, and C. glabrata ATCC 39316 were from the American Type Culture Collection (ATCC, Manassa, VA, USA), Cryptococcus neoformans IFM 5844 and IFM 5855 were from IFM Quality Services Pty Ltd (IFM, Ingleburn, NSW, Australia) and Aspergillus fumigatus SzMC 2486, A. flavus SzMC 2536, A. niger SzMC 2761, A. terreus SzMC 1932 were from Szeged Microbiological Collection (SzMC, Szeged, Csongrad, Hungary). In order to check whether the method we developed is generally applicable, non-human pathogenic fungal strains F. oxysporum SzMC 0609 and M. hiemalis SzMC 0478 were also included in the study. Furthermore, to confirm the reliability and reproducibility of the technique, clinical strains of C. albicans (n=14), C. glabrata (n=5), C. tropicalis (n=4), C. parapsilosis (n=5), C. krusei (n=4), C. quillermondii (n=4), C. lusitaniae (n=3), C. norvegensis (n=1), C. inconspicua (n=2), C. dubliniensis (n=2) and Cryptococcus neoformans (n=2) from the Institute of Clinical Microbiology at the University of Szeged were also tested. These strains were maintained on Sabouraud – chloramphenicol slant agar and periodically sub-cultured. 24-h cultures were used for all subsequent experiments. The species identities of the clinical isolates were confirmed by conventional biochemical methods.
DNA extraction. DNA extraction was carried out as reported by Liu et al. (25), with a slight modification: in the first step, a modified lysis buffer [50 mM Tris-HCl (pH 8.0), 1 mM EDTA (pH 8.0), 28 mM β-mercaptoethanol] supplemented with 1% Helix pomatia gastric juice was applied. The incubation time, temperature and other conditions of the procedure were as in the original protocol.
To determine the sensitivity of the reaction, 300 μl of human blood were infected by C. albicans reference strain, and used for DNA extraction. The diluted DNA samples were then used for PCR reaction. The number of fungal cells was determined by plating the aliquots of serially diluted samples onto Sabouraud-glucose agar.
Primer selection. The panspecific fungal primers designed to amplify the ITS2 regions of fungal ribosomal DNA (28) were as follows: ITS86 (gTg AAT CAT CgA ATC TTT gAA C) as forward and ITS4 (TCC TCC gCT TAT TgA TAg C) as reverse primer. The sizes of the amplified PCR products ranged between 200 and 500 base pairs (bp), depending on the fungal strain.
PCR reaction. A Bio Rad CFX instrument (Bio-Rad Laboratories, Hercules, CA, USA) was used for amplification. The reaction volume of 10 μl contained 1 μl of fungal DNA, 1.0 μM of each of the primers and 5 μl of reaction buffer, which includes EvaGreen dye (SsoFast Supermix; Bio-Rad). The PCR conditions were as follows: initial denaturation at 95°C for 420 s, followed by 40 cycles of denaturation (95°C for 30 s), annealing (55°C for 60 s) and extension (72°C for 60 s). The melting curve analysis at the end of the amplification consisted of one cycle starting at 72°C for 20 s, the temperature subsequently being increased to 95°C in 0.1°C/s increments (HRM analysis). The Bio Rad CFX Manager Software version 1.6 and Precision Melt Analysis Software 1.1 (Bio Rad) were used for data evaluation.
LightCycler DNA master SybrGreen I (Roche Diagnostics GmbH, Mannheim, Germany) mastermix (MMX), Maxima qPCR SybrGreen no ROX (Fermentas Inc., Vilnius, Lithuania) MMX and the IQ SybrGreen Supermix (Bio Rad) MMX were also used for melting point investigations according to the manufacturer's instructions.
For comparison, the SybrGreen melting peak data were measured with the LightCycler real-time instrument (Roche Diagnostics GmbH), as described previously (22).
Results
DNA samples from all the fungal species studied were prepared and amplified successfully with the EvaGreen dye-based method in the Bio Rad CFX instrument. Species-specific melting peaks (Tm) were obtained via the HRM analysis, allowing the differentiation of all the investigated fungal species. Thus, it was possible to distinguish the 15 most common fungal pathogens. The clinical strains had the same Tm as the references and were used in the calculation of the standard deviation (SD). Due to the highly saturating EvaGreen dye and the HRM analysis, the accuracy of the resolution was ±0.01–0.24°C. The sensitivity of the reaction was 5 colony-forming units (cfu)/reaction using the C. albicans reference strain.
For comparison, all the fungal strains were investigated with the SybrGreen dye and the Roche LightCycler instrument. The sensitivity of the reaction was similar, but the SD was higher, at ±0.02-1.04°C. The Tm data for the fungal strains are listed in Table I.
The determination of Tm is very sensitive to the composition of the PCR reaction mixture, and primarily to the ionic strength. To avoid Tm bias between PCR runs originating from pipetting errors, the application of MMX is advisable. One limitation of this method is that the various forms of MMX offered by different suppliers differ in reagent composition, which may also influence the Tm values. For example, the difference in Tm between C. albicans and C. dubliniensis was earlier determined with the LightCycler DNA master SybrGreen I MMX to be 1.2°C (87.4 and 86.2°C) (29). With the Maxima qPCR SybrGreen no ROX MMX, the difference was 2.0°C (83.5 and 81.5°C), with the IQ SybrGreen Supermix it was 1.5°C (85.6 and 84.1°C) and with the SsoFast Supermix it was 1.0°C (84.5 and 83.5°C). The situation is similar for other fungi (data not shown). Naturally, repeated runs with a given MMX lead to reproducible data. In the event of a change of MMX, ‘calibration’ is necessary to establish the new Tm data on the fungal strains.
The data determined in the present work were obtained with the use of SsoFast Supermix.
Discussion
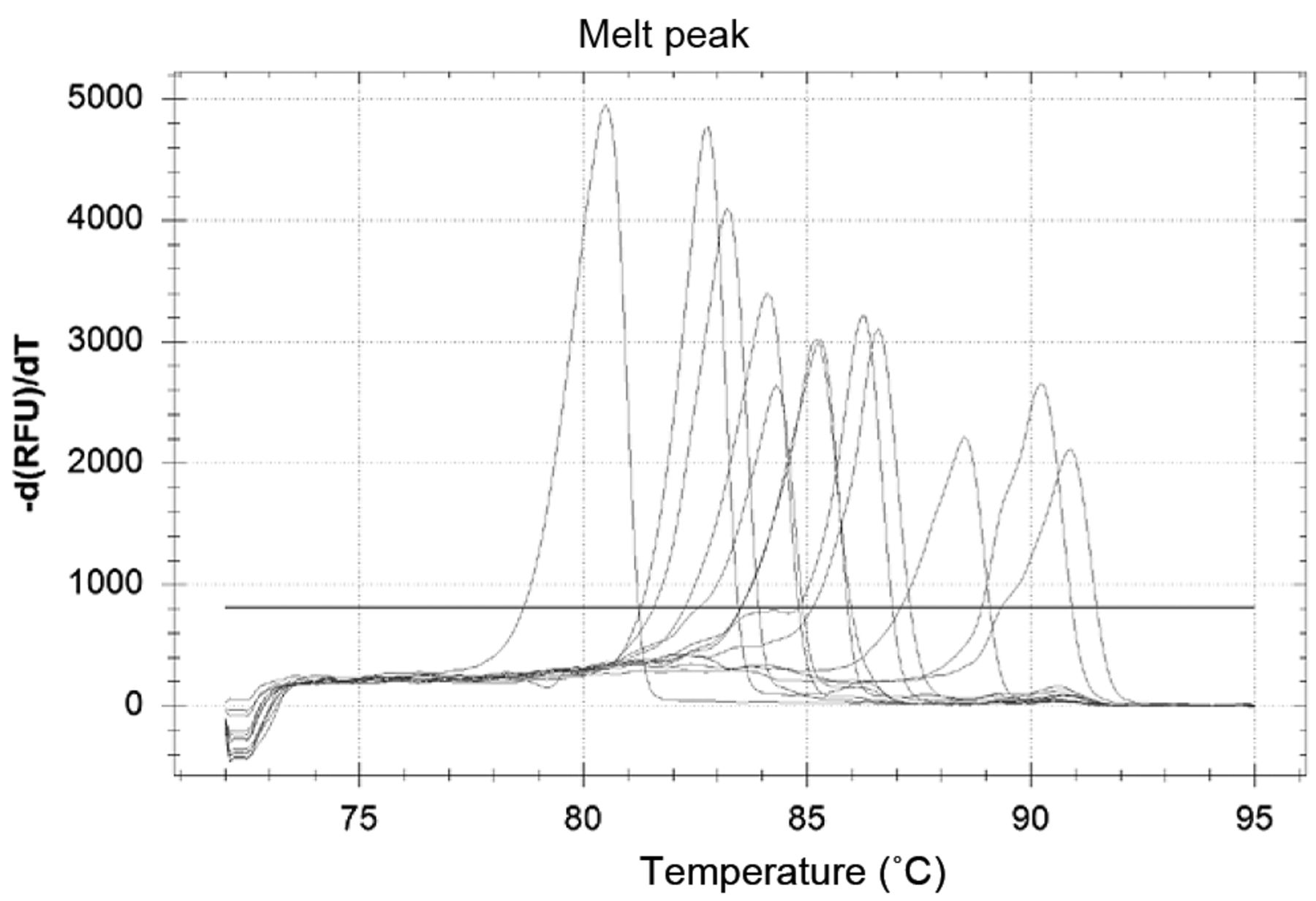
In our experimental set-up, real-time PCR was chosen to establish a melting-point analysis-based diagnostic method for the fungal species that are the most frequent causative agents of invasive fungal infections. Real-time PCR is currently one of the fastest diagnostic methods (26). The use of rDNA genes for the detection is based on the conserved sequences of 5.8S and 28S rDNA. There are no differences in sensitivity (27), only in amplicon length (28), between the previously described primer pairs amplifying this region. Accordingly, it is possible to differentiate between the fungal species by electrophoresis (12, 28), or by melting analysis (22, 23, 29). The Roche LightCycler PCR was specially constructed to amplify amplicons under 500 bp. The amplicons amplified by the ITS86/ITS4 primer pair range between 192 bp (Geotrichum candidum) and 494 bp (Malassezia furfur), which is perfectly suitable for this instrument profile. The same primers were used in our new HRM examinations because the shortest amplicons are more sensitive to the sequence alterations than are the longer ones. High-resolution software is used for the detection and evaluation of mutations and polymorphisms in the short amplicons. In this context, users apply positive controls to compare unknown PCR amplicons with chosen genotypes. This method can readily be applied to distinguish certain genetic alterations. The differentiation of the investigated fungal strains with the HRM software is more difficult. The software investigates the relative ratios between the fluorescence curves, and thus all of the expected fungal species have to be used as positive controls in every PCR run (Figure 1). The melting peaks offer absolute values for the differentiation. The literature reports indicate that in the case of yeasts, 99% of fungal septic infections are caused by eight species: C. albicans, C. tropicalis, C. glabrata, C. parapsilosis, C. krusei, C. guilliermondii, C. lusitaniae and Cryptococcus neoformans (30, 31). These most common fungal pathogens can be differentiated with the help of these melting peak differences (Figure 2). Furthermore, Aspergillus species can also be detected with this primer pair. The Tm values of the common fungal pathogens A. fumigatus, A. flavus and A. niger are different and their discrimination both from Candidas and from each other is possible (Table I).
Comparison of melting points. The previous studies used the Roche LightCycler, SybrGreen dye and melting analysis. The present data were measured by means of the Bio-Rad CFX Polymerase chain reaction machine, EvaGreen dye and High resolution melting analysis.
Additionally, we determined the Tm values of filamentous fungi which are non-pathogenic (M. hiemalis) or very rarely pathogenic (F. oxisporum) to humans. M. hiemalis is easily distinguished, but F. oxysporum has same the Tm as that of C. lusitaniae. This illustrates that although this method is able to differentiate the most common fungal pathogens, it is not suitable as a general method of fungal species identification.
The fungal load in fungal sepsis is generally below 10 cfu/ml (18). As the sensitivity of this PCR is 5 cfu/reaction, in combination with a correct preparation method, it is suitable for the detection of invasive Candida infections.
The incidence of fungaemia has increased over the last decades (1). The early detection of fungal pathogens has a great impact on the clinical outcome of the infection. We have improved the panfungal PCR by means of the HRM investigation, thereby achieving the distinction of 99% of fungal pathogens in sepsis. Thus, the protocol furnishes an opportunity for the rapid detection and reliable differentiation of the Candida and Aspergillus species most frequently isolated from clinical samples.
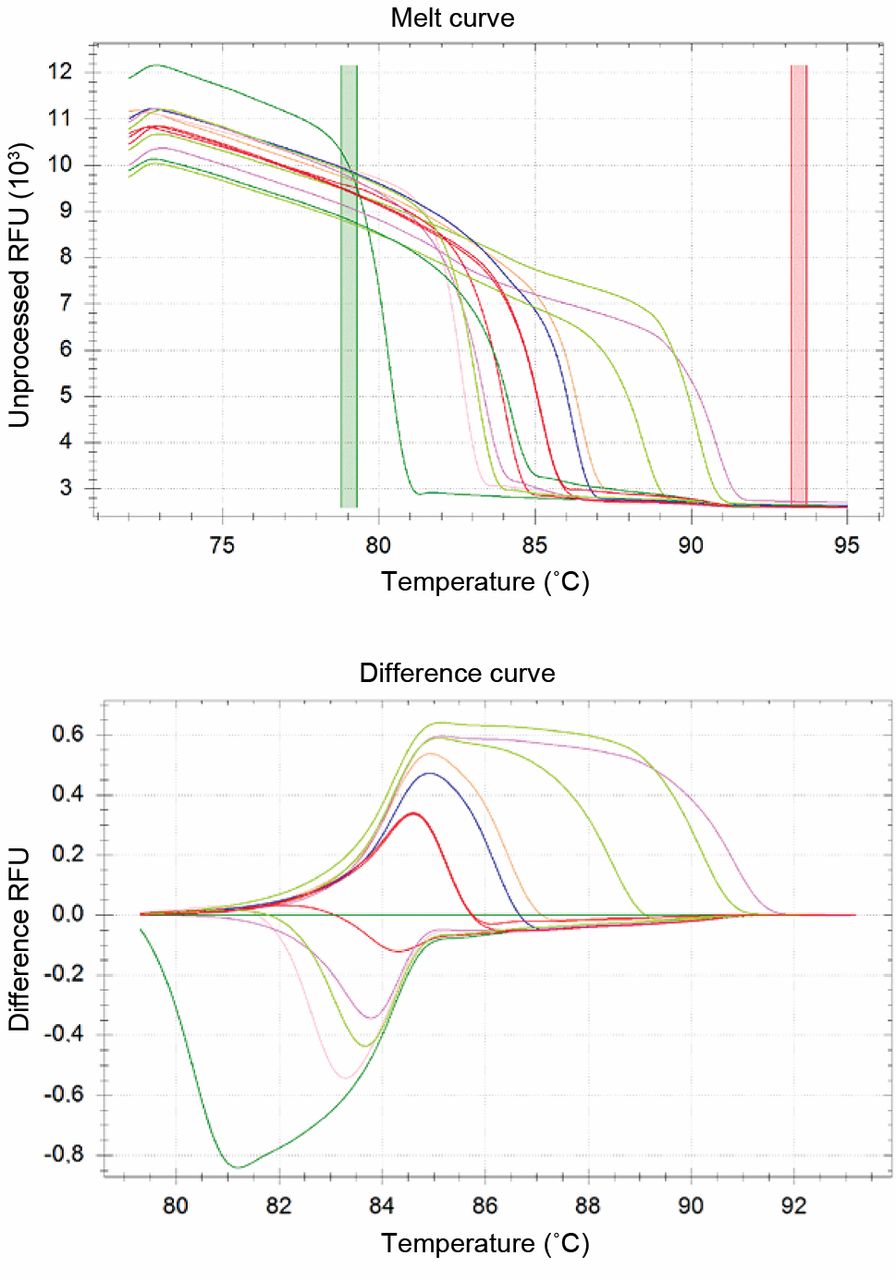
Differentiation of the fungal strains with the software. a: The descending fluorescent signals. The temperature was increased to 95°C after the last step of the PCR reaction and the fluorescence signal was monitored continuously during the temperature ramp. The relative fluorescence units (RFU) were plotted against the temperature. The coloured vertical stripes are the variable baselines for the HRM calculation. Fungal strains from left to right: Mucor hiemalis; C. quillermondii; C. glabrata; C. dubliniensis; Cryptoc. neoformans; C. albicans; C. lusitaniae and F. oxysporum together (red lines); C. norvegensis; C. inconspicua; C. krusei; A. flavus and A. fumigatus. b: The HRM analysis. The HRM software calculated the relative ratio between the melting temperatures. The user option is to choose the basic curve/genotype for comparison. Here the C. albicans was used (dark green line in the centre).

{kind=link}
{kind=link}
Differentiation of the fungal strains via the melting peaks. The descending fluorescence signals were transformed to derivative melting curves [-d(RFU)/dT]. These peaks allow for absolute discrimination between fungal strains. The order of the fungal strains is the same as the one given in the legend to Figure 1.
Acknowledgements
This work was supported by the project “TÁMOP-4.2.1/B-09/1/KONV-2010-0005 – Creating the Center of Excellence at the University of Szeged” is supported by the European Union and co-financed by the European Regional Fund and the Foundation for Cancer Research Szeged (Hungary).
- Received May 11, 2012.
- Revision received September 14, 2012.
- Accepted September 17, 2012.
- Copyright © 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved