

Abstract
Tetrahymena thermophila is a unicellular eukaryotic model organism used for a variety of biochemical, molecular and biological studies. According to its macronucleus genome sequence, it is expected to contain more than 27,000 protein-coding genes, although only a small proportion of them have information published specifically about them. Here, we present a reference map for whole cell lysate of T. thermophila obtained using two-dimensional gel electrophoresis (2-DE) combined with mass spectrometry. Although (2-DE) is one of the most efficient techniques for resolving complex protein mixtures and revealing the relative high-abundance proteins, it has not yet been applied generally to ciliates. In order to obtain qualitative protein samples for analysis, an appropriate homogenization method is required. Optimization of the homogenization method led to the analysis of nearly 4500 protein spots, the final identification of 375 different proteins using Mascot software and an additional 258 gene products using a newly developed web service, called Peptide Finder, resulting in a total of 631 different gene products that are considered to constitute the proteomic profile of the whole cell lysate of T. thermophila.
- MALDI-TOF
- proteome
- proteomics
- Tetrahymena thermophila
- TRI reagent
- 2-D electrophoresis
- Peptide Finder
Tetrahymena thermophila is a unicellular eukaryotic organism that belongs to the division of protists. It is a non-pathogenic, aerobian that possesses characteristic structures, such as cilia. Due to its structural and functional complexity, as well as the fact that its genome preserves many primary biochemical procedures of eukaryotes (1), T. thermophila is considered to be a valuable model organism for genetic and biological studies (2, 3). Its use has led to the discovery of important biomolecules such as catalytic RNA, telomere, telomerase and the cell motor dynein, as well as elucidation of the role of histone acetylation in gene expression (4). Lately, T. thermophila has been widely used for enzyme purification and in bioreactors (5-8).
Like many ciliates, T. thermophila possesses two types of nuclei, distinct in their function (9). The micronucleus (MIC) contains five pairs of mostly transcriptionally silent chromosomes, while the macronucleus (MAC) determines the phenotype of the cell. The genome of the latter has only recently been annotated and found to consist of 225 transcriptionally active chromosomes with a total size of 104 Mb; 27,000 protein-encoding sequences have been identified (10). The mitochondrial proteome of T. thermophila was recently published (11), while from previous studies, data about its ciliome (12), its phagosomic proteins (13), basal body proteins (14) and four novel β/γ crystalline domain proteins from a subcellular fraction, enriched in granules (15) were made available. Out of the 27,000 predicted protein-encoding sequences, approximately 15,000 have strong BLAST matches to known or predicted genes from other organisms. However, only 53 proteins are annotated in the protein database of UniProtKB/Swiss-Prot and 17,715 in UniProtKB/TrEMBL. In the present work, we applied proteomic technologies coupled with mass spectrometry and bioinformatics approaches in order to characterize the proteome of T. thermophila and construct its reference map.
Materials and Methods
Cell cultures. T. thermophila was cultured at 25°C under aerobic conditions in a medium consisting of 2% (w/v) proteose-peptone, 0.5% (w/v) sucrose, 0.2% (w/v) yeast extract and 1% (v/v) Fe2+ -9 mM EDTA (pH 5.5) (16). At the end of the logarithmic phase (~72 h), cells were harvested, centrifuged and the cell pellet was washed using 0.9% NaCl.

Peptide map of total protein extract from T. thermophila. The first dimensional separation was conducted in a 3-10 immobilized pI gradient strip. The identified proteins are annotated by their accession numbers as listed in Table I.
Homogenization. Protein extraction was achieved using TRI-Reagent (17), an organic solution of phenol and guanidine thiocyanate that facilitates protein extraction with the simultaneous removal of DNA and RNA content. DNA and RNA are endogenous charged particles that enclose proteins and inhibit them from being separated during two-dimensional gel electrophoresis (2-DE). The use of TRI Reagent not only facilitates cell homogenization due to its organic compounds, but enriches the sample's protein content as it removes inhibiting molecules, giving clear, highly reproducible gel images and identifications.
Cells from cell cultures were homogenized in TRI Reagent as recommended by the manufacturer (Ambion/Applied Biosystems, Austin, TX, USA). A total of 10×106 cells were homogenized in 1 ml TRI reagent and incubated for 5 min at room temperature. The solution was centrifuged at 12000×g for 5 min to remove undiluted material and 200 μl chloroform were added to the supernatant. After incubation for 15 min and centrifugation at 12000×g for 10 min, the RNA phase was removed and 300 μl ethanol were added to the residual phenol phase and interface to sediment the DNA. After 2-3 min, the solution was then centrifuged at 2000×g for 5 min. Three volumes of acetone were added to the supernatant, incubated for 10 min at room temperature and centrifuged at 12000×g for 10 min.
The protein pellet was washed with 1 ml of protein Wash 1 buffer (300 mM guanidine hydrochloride in 95% ethanol, 2.5% glycerol (v/v)) and incubated for 10 min at room temperature, centrifuged at 8000×g for 5 min, washed twice more with 1 ml of Wash 1 buffer and then washed with Protein Wash 2 (ethanol containing 2.5% glycerol (v/v)) for 10 min.
Two-dimensional electrophoresis (2-DE). Protein pellets were pooled and resuspended in urea lysis buffer (20 mM Tris, 7 M urea, 2 M thiourea, 4% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 10 mM 1,4-dithioerythritol, 1 mM EDTA) and a mixture of protease inhibitors [1 mM PMSF and 1 tablet complete™ (Roche Diagnostics, Basel, Switzerland), per 50 ml of suspension buffer] and phosphatase inhibitors (0.2 mM Na2VO3 and 1 mM NaF). The protein content in the supernatant was determined by applying the Bradford method (18) using BIO-RAD protein assay (BIO-RAD Laboratories, Hercules, CA, USA). 2-D Gel electrophoresis was performed as previously reported (19). Samples of 1.0 mg total protein were applied on immobilized 3-10 pI, 4-7 pI and 6.3-8.3 pI non-linear gradient strips (17 cm) at their basic and acidic ends. Focusing for the former two strips started at 250 V for 30 min and the voltage was gradually increased to 5000 V at 3 V/min and remained constant for an additional 16 h. For pI 6.3-8.3, strip rehydration lasted only 6 hours and focusing started at 250 V for 30 min and the voltage was gradually increased to 8000 V at 3 V/min and remained constant for an additional 15 h.
The second-dimensional separation was performed in 12% SDS-polyacrylamide gels (180×200×1.5 mm), running at 40 mA per gel in a PROTEAN apparatus (BIO-RAD). After fixation with 50% methanol containing 10% acetic acid for 2 h, the gels were stained overnight with colloidal Coomassie blue (Novex, San Diego, CA, USA), washed twice with water and scanned in a densitometer (GS-800 Calibrated Densitometer; BIO-RAD).
Peptide mass fingerprint and post source decay. Peptide analysis and protein identification were performed as previously described (20). Spots were detected using Melanie 4.02 software (GeneBio, Geneva Swiss) on the Coomassie blue-stained gel and the spots were then excised by Proteineer SPII (Bruker Daltonics, Bremen, Germany), destained with 30% acetonitrile in 50 mM ammonium bicarbonate and dried in a speed vacuum concentrator (MaxiDry Plus; Heto, Allered, Denmark). Each dried gel piece was rehydrated with 5 μl of 1 mM ammonium bicarbonate, containing 50 ng trypsin (Roche Diagnostics) and left for 16 h at room temperature. A total of 10 μl of 50% acetonitrile, containing 0.3% trifluoroacetic acid, were added to each gel piece and incubated for 20 min with constant shaking. A peptide mixture (1.5 μl) was simultaneously applied with 1 μl of matrix solution, consisting of 0.025% α-cyano-4-hydroxycinnamic acid, CHCA (Sigma-Aldrich, St. Louis, MO, USA) and the internal standard peptides [Des-Arg]-bradykinin (904.4681 Da; Sigma) and adrenocorticotropic hormone fragment 18-39 (2465.1989 Da; Sigma) in 65% ethanol, 35% acetonitrile and 0.03% trifluoroacetic acid.
Samples were analyzed for peptide mass fingerprint (PMF) with matrix assisted laser desorption ionization – Mass Spectrometry (MALDI-MS) in a time-of-flight mass spectrometer (Ultraflex II MALDI-TOF-TOF MS/MS; Bruker Daltonics, Bremen, Gernamy). Peptide matching and protein searches were performed automatically, as described by Berndt et al. (20). Each spectrum was interpreted by the Mascot Software (Matrix Sciences Ltd., London, UK) and Peptide Finder. For peptide identification, the monoisotopic masses were used and a mass tolerance of 0.0025% was allowed. Unmatched peptides or peptides with up to one miscleavage site were not considered. The peptide masses were compared with the theoretical peptide masses of all available proteins from all species using Swiss-Prot and TrEMBL databases. The probability score identified by the software was used as the criterion of the identification (http:/www.matrixscience.com). Samples not identified by PMF (probability significance of p<0.05) were automatically selected for post-source decay (PSD) MS-MS analysis with MALDI-MS-MS in the same spectrometer. The peptide masses chosen for PSD-MS-MS analysis had a signal intensity of >600 counts and were excluded from the trypsin autodigest, matrix and keratin peaks. The resulting PSD spectra were also interpreted by the Mascot Software and Mascot probability-based scores of p<0.02 were considered significant.
{kind=link}
{kind=link}
The identified proteins were automatically annotated on the gel image by ProteinScape software (Bruker Daltonics). Obtained spectra did not result in reliable significance in protein identification through the Mascot software, re-evaluated with a newly developed web service called Peptide Finder (21). The search was carried out through a database of completely digested T. thermophila proteins (http://bioserver-1.bioacademy.gr/Bioserver/PeptideFinder/). For the identification, complete peak lists with measured molecular weights obtained by the MALDI-MS analysis were uploaded to the software and mapping to the proteins of the database requested. Additionally, various filters are available in order to obtain a more refined list of peptides and proteins with the requested molecular weights. Peptide Finder Score is an index used to comparatively rank the candidate proteins based on the statistics of the molecular weight distributions. It suggests that a good identification should be based on the number of molecular mass matches, the frequency they present inside the Swiss-Prot database for the species (i.e. their randomness index) and the molecular mass (indicating the size) of the suggested protein.
Results
The objective of the present study was to characterize the relevant proteins of high and medium abundance of T. thermophila homogenate. For this reason, we employed 2-D gel electrophoresis technique. The homogenates were analysed in three different pH ranges (3-10, 4-7, 6.3-8.3) and 4,457 spots were totally detected by the Melanie 4.02 software. These spots were excised from the gels and analyzed for protein identification following in-gel digestion with trypsin. Each spot was analyzed for PMF with MALDI-MS in a time-of-flight mass spectrometer and proteins were identified automatically by the peptide mass matching. Proteins not identified by PMF were subsequently selected for PSD-MS-MS and analyzed with MALDI-MS-MS. Using an internal peptide standard to correct the measured peptide masses, we were able to use very narrow windows of mass tolerance (0.0025%) and hence, increase the confidence of identification, as well as the total identification rate up to 85%. This procedure resulted in the identification of 375 different gene products (Figure 1). The abbreviated and full names of the proteins, the theoretical MW as well as data from the MS analysis, such as the probability that the identification is a random event (Score), and the coverage of the protein by the identified peptides are listed in Table I. The protein identification data are available at the PRIDE database (http://www.ebi.ac.uk/pride/, experiment #3700). It should be noted that many of the identified proteins in these gels were represented by more than one spot with the same apparent molecular mass, but with different pI values, possibly due to post-translational modifications or isoenzyme variations.
Proteins from T. thermophila were extracted and separated by 2-D gel electrophoresis as described in the Materials and Methods. Proteins were identified by MALDI-MS and MS/SM, following in-gel digestion with trypsin. The proteins identified by Mascot Software are designated by their Swiss-Prot accession numbers and their full names. The theoretical MW as well as the protein amino acid sequence coverage by the matching peptides are given as an indication of the confidence of the identification. Mascot Score is −10 log(P), where P is the probability that the observed match is a random event.
Proteins from T. thermophila were extracted and separated by 2-D gel electrophoresis as described in the Materials and Methods. Proteins were identified by MALDI-MS and MS/SM, following in-gel digestion with trypsin. The proteins identified by the PeptideFinder software are designated by their SWISS-PROT accession numbers and their full names. The theoretical MW as well as the protein amino acid sequence coverage by the matching peptides are given as an indication of the confidence of the identification. PeptideFinder score is an index used to comparatively rank the candidate proteins based on the statistics of the molecular weight distribution.
Mass spectra that did not result in protein identification by the above procedure were processed for identification by the Peptide Finder software (21). By that procedure, we were able to identify 258 additional gene products (Table II), resulting in a total of 631 proteins that are considered to make up the proteome of T. thermophila.
Discussion
T. thermophila is one of the most studied model organisms in biology. Although numerous studies concerning specific proteins or subsets of proteins after fractionation exist, to date nothing has been published regarding its whole proteome. This is mainly due to the fact that T. thermophila has a very large number of protein-encoding sequences to be analysed all at once, with a single technique. Each technique has its limitations and requires compatibility of the homogenization and identification methods. In this study, we were able to identify a significant number of proteins, but only after proper optimization of the homogenization method, namely, by the removal of nucleic acids with the use of Tri-Reagent for the protein isolation instead of the conventional method (urea-thiourea) (data not shown).
Out of the 631 identified proteins, 8 have already been annotated in the Swiss Prot database, indicating that the remaining 623 proteins are as yet considered to be hypothetical. This is because Swiss Prot is a protein database that contains only proteins actually identified and studied, while TrEMBL is generated by computer translation of the genetic information from the EMBL Nucleotide Sequence Database. As expected, the latter procedure is subject to errors, so proteins predicted by the TrEMBL database have a risk of being hypothetical. The identification of these proteins and thus, the confirmation of a significant number of proteins found in the TrEMBL database all at once, was a major achievement.
A total of 92% of the identified proteins are correlated to the genome of T. thermophila, while the remaining 8% show high homology with proteins of other protozoa such as Stylonychia, Gastrostyla, Frontonia, Strombidinopsis, Toxoplasma, Plasmodium, Paramecium, Cryptosporidium, Theileria, Favella, and Chilodonella.
A brief comparison between our work and other studies, revealed only few similarities. Out of the 631 identified proteins, only 11 are common to those published for its ciliome (12) and 34 proteins are found in both our results and those for the mitochondrial proteome (11). As to be expected, this is a result of the different preparation and identification techniques used and mainly due to the fact that we chose to analyze the entire homogenate of the organism.
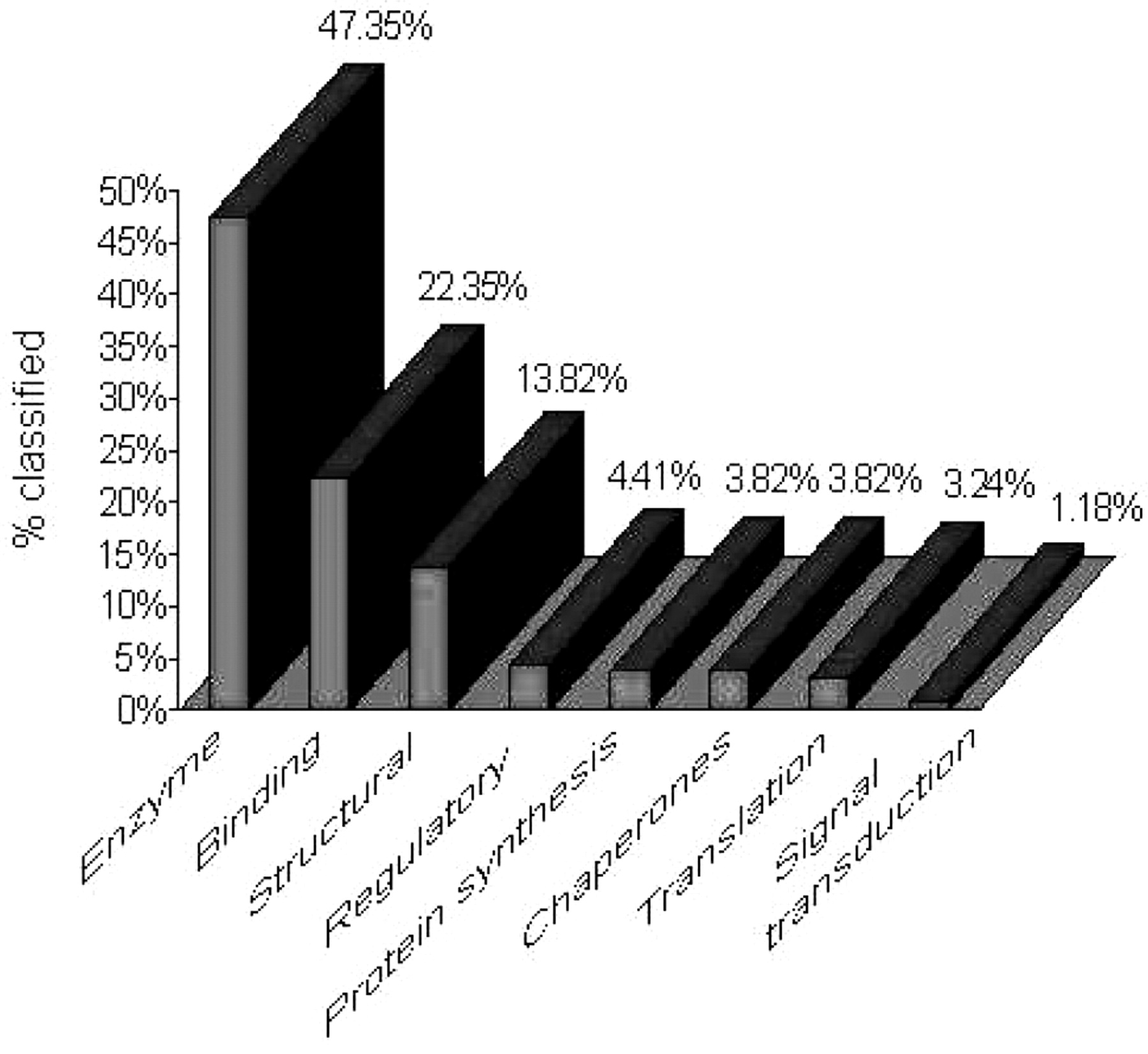
Nearly half of these identified proteins have unknown functions, while of the rest 47.5% are enzymes (lyases, ligases, isomerases, proteases, hydrolases, etc.) participating in various metabolic reactions, 22.4% are proteins that bind other molecules and ions for their action, such as ATP-, GTP-RNA and calcium-binding proteins (Figure 2). A total of 13.8% correspond to structural proteins, such as actin, tropomyosin, centrin, dynein and Granule lattice proteins, 4.4% were regulatory proteins, such as 14-3-3, AhpC/TSA and N-ethylmaleimide-sensitive factor and 3.8% are ribosomal proteins involved in protein synthesis. Another 3.8% are proteins chaperones, such as heat-shock proteins 90, 82 and DNAK, and 3.2% are translational proteins, such as translation elongation factor ef-1. Finally, 1.2% of the identified proteins are involved in signal transduction pathways, as the Mov34/MPN/PAD-1 and Ras protein families.
The identified proteins are considered to be the most abundant (22), but this number may easily seem small for the entire homogenate of such a complicated organism. However, since no further steps have been taken in proteomics for the analysis of total cell extract of T. thermophila and since protein databases are continuously altered, as they are based on the translation of the mRNA without taking into account the alterations that may occur before the final product of protein is reached, their addition to the list of proteins found to be expressed from T. thermophila is a notable contribution to the study of this organism.
The obtained results indicated that even though most of the identified proteins are essential for several functions of this protozoon and participate in various metabolic reactions, there is still a significant percentage corresponding to proteins of unknown function. The future aim is to elucidate their role or even suggest a possible connection with the existing functions. This indicates that Tetrahymena is an organism to be studied for many years and should still be a reliable model organism for biochemical and biological studies.
- Received October 14, 2009.
- Revision received April 14, 2010.
- Accepted April 23, 2010.
- Copyright © 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved