


Abstract
Background/Aim: We examined the mechanism of nitric oxide (NO) production in a tissue-factor (TF)-induced disseminated intravascular coagulation (DIC) model in rats, using inducible nitric oxide synthase (iNOS) inhibitor (L-NIL), endothelial nitric oxide synthase (eNOS) inhibitor (L-NAME), Factor Xa inhibitor (DX-9065a), and thrombin inhibitor argatroban. Materials and Methods: Experimental DIC was induced by sustained infusion of 3.75 U/kg TF for 4 h via the tail vein. We then investigated the effect of these four agents on TF-induced DIC. Results: Administration of L-NIL or L-NAME during induction of TF-induced DIC did not affect hemostatic markers, whereas elevated plasma levels of NO metabolites (NOX) were significantly suppressed by co-administration of L-NAME. A significant increase in eNOS-mRNA expression was observed in the TF-induced DIC model. Argatroban almost completely suppressed eNOS-mRNA expression. Conclusion: eNOS plays an important role in the NO production in the TF-induced DIC, and thrombin is a key stimulant of eNOS-mRNA expression in this model.
- Disseminated intravascular coagulation
- nitric oxide
- endothelial nitric oxide synthase
- inducible nitric oxide synthase
Disseminated intravascular coagulation (DIC) represents a serious condition in which activation of coagulation and promotion of fibrinolysis in the circulating blood occur simultaneously (1, 2). Organ failure and bleeding are characteristic symptoms of DIC (3, 4). Disturbances of microcirculation due to the formation of numerous microthrombi in various organs cause multiple organ failure (MOF) (5, 6). It has been reported that vasoactive substances play an important role in the pathophysiology of DIC (7-9).
Nitric oxide (NO) is a typical messenger involved in vasodilation. There are two types of isoforms of NO synthase. NO is synthesized by the endothelial isoform NO synthase (eNOS) in normal endothelial cells and by the inducible isoform NO synthase (iNOS) in endothelial cells stimulated by cytokines or lipopolysaccharide (LPS) (10-12). iNOS-mediated enhancement of NO synthesis occurs during septic shock and cytotoxicity (13). NO has an inhibitory effect on thrombus formation, suppressing the production of tissue factor (TF) and plasminogen activator inhibitor (PAI) in vascular endothelial cells (14-18).
We previously clarified the role of vasoactive substances, such as NO and endothelin-1 in the development of DIC, using TF-induced and LPS-induced DIC models in rats (19). Plasma levels of NOX (metabolites of NO) were significantly increased as a result of LPS- or TF-induced DIC in rats. Excess NO production due to enhancement of iNOS activity is well-documented in LPS-induced sepsis, although little is known about NO production during DIC (20, 21). To the best of our knowledge, we are the only group that has reported excess NO production in a model of TF-induced DIC (19), although it is unclear which isozyme is involved in this process.
Here, we examined the mechanism of NO production in a TF-induced DIC model in rats, using a selective iNOS inhibitor [N (6)-(iminoethyl)-lysine (L-NIL)] (22, 23), a relatively selective eNOS inhibitor [L (omega)-nitro-L-arginine methylester (L-NAME)] (24), a specific Factor Xa inhibitor (DX-9065a) (25), and a specific thrombin inhibitor (argatroban) (26).
Materials and Methods
Animals. Animals were maintained according to the Standards of Animal Care and Experimentation published by our institution. All animal experiments were approved by the Committee on Animal Experimentation of Kanazawa University (AP-183920).
Male Wistar rats (aged 6-7 weeks and weighing 160-170 g) obtained from Nippon SLC (Hamamatsu, Japan) were acclimatized for at least 3 days in our animal quarters before experimentation.
General experimental procedures. Blood was withdrawn from the abdominal aorta of rats anesthetized with pentobarbital sodium (Nembutal, 30 mg/kg i.p.) into plastic syringes 4 and 8 h after TF administration. All samples were diluted (1:9 v/v) with 4% sodium citrate.
The control group was administered a sustained infusion of 10 ml physiological saline for 4 h via the tail vein. Blood was withdrawn 4 and 8 h later (n=6 for both groups). None of the parameters examined demonstrated significant changes during the study (data not shown).
TF-induced experimental DIC (TF group). Thromboplastin (Thromboplastin C, Dade Diagnostics of P.R. Inc., Aguada, Puerto Rico, USA) (containing rabbit brain thromboplastin and CaCl2) was dissolved in physiological saline immediately before use. Experimental DIC was induced by sustained infusion of 3.75 U/kg thromboplastin diluted in 10 ml of saline for 4 h (n=6) into the tail vein. We have already confirmed that this method of TF administration does not change plasma calcium levels in rats.
Effect of concurrent drug administration on TF-induced experimental DIC. We administered L-NIL (6 mg/kg) (Sigma Aldrich, St. Louis, MO, USA), L-NAME (20 mg/kg) (Sigma Aldrich), argatroban (AR: 2 mg/kg) (Sigma Aldrich), and DX9065a (DX: 2 mg/kg) (Daiichi Pharmaceutical Co., Ltd., Tokyo, Japan) to rats 30 min prior to TF infusion followed by continuous infusion of each drug for a further 4 h (n=6 per group). Each drug was diluted in 1 ml of saline for the first 30 min of infusion, after which each drug was diluted in 10 ml of saline for simultaneous infusion with TF for the next 4 h.
Administration of individual drugs alone. The control groups for each agent were administered a sustained infusion of drug diluted in 11 ml of physiological saline for 4.5 h via the tail vein (n=6 per group). In all groups, except for the L-NAME group, none of the parameters examined demonstrated significant changes at 4 or 8 h (data not shown). In the group receiving L-NAME alone, prothrombotic tendency was induced (previously published data) (27).
Parameters. Platelets (PLT) were counted using an automated device for animals (Celltac α, MEK-6558, Nihon Kohden Co., Tokyo, Japan) within one hour of sampling. Citrated plasma samples obtained by whole blood centrifugation were stored at -80°C until analysed. Plasma fibrinogen levels were determined by clotting assay. D-dimer levels were determined using the quantitative latex agglutination test (ELPIA ACE DD dimer, LSI medience, Tokyo, Japan). Plasma thrombin-antithrombin complex (TAT) levels were determined using a commercial enzyme-linked immunosorbent assay (ELISA) kit (Enzygnost TAT, Behringwerke, Marburg, Germany).
Plasma levels of NOX (nitrite/nitrate; NO2/NO3), which are the final products of NO in vivo, were determined using a colorimetric assay kit (Nitrate/Nitrite assay kit, Cayman Chemical, Ann Arbor, MI, USA).
Reverse transcriptase-polymerase chain reaction assay. Another experiment was performed to investigate the mRNA expression of eNOS and iNOS in the control group, the LPS group (positive control group for iNOS-mRNA expression), the TF group, and the TF+argatroban group (n=4 for each group). Apart from the LPS group, the same method of drug administration described above was used for each group. In the LPS group, LPS (Escherichia coli 055: B5 lipopolysaccharide B; Sigma Aldrich) was freshly dissolved in physiological saline before each experiment. Experimental DIC was induced by sustained infusion into the tail vein of 30 mg/kg LPS diluted in 10 ml of saline over 4 h.
For RNA extraction, the lungs were quickly removed at 4 h. Total RNA was prepared from the lung samples with the Absolute RNA Microprep Kit (Agilent, Santa Clara, CA, USA). Total RNA was reverse-transcribed into cDNA in a reaction primed with oligo (dT) using SuperScript II reverse transcriptase, as recommended by the manufacturer (Thermo Fisher Scientific, Waltham, MA, USA). The primer sequences for β-actin and NOS (Amersham Pharmacia Biotec, Amersham, UK) are described here. β-actin, forward: 5’-TCC TAC AAT GAG CTG CGT GTG GC-3’, reverse: 5’-CTC(A/G)TA GCT CTT CTC CAG GGA GGA-3’. i-NOS, forward: 5’-ACA TCC TGC AGA AAG AGC TG-3’, reverse: 5’-TCA GAG TCT TGT GCC TTT GG-3’. e-NOS, forward: 5’-ACT GGC ATT GCA CCC TTC CG-3’, reverse: 5’-GTT GCC AGA ATT CTC TGC AC-3’. PCR cycling was performed as follows: denaturation for 30 s at 94°C, annealing for 1 min at 58°C (β-actin) or for 30 s at 53°C (eNOS and iNOS), extension for 1.25 min at 72°C (β-actin) or for 30 s at 72°C (eNOS and iNOS). PCR was completed after 22 cycles for β-actin and 35 cycles for eNOS and iNOS. PCR products were separated by 2% agarose gel electrophoresis and stained with ethidium bromide (EtBr). Only PCR products with a distinct target band corresponding to an appropriate product on the electrophoresis gel were used for further analysis. Fluorescence was measured with a UV-Transilluminator (AB-1500, ATTO., Tokyo, Japan). The relative amount of eNOS, iNOS, and β-actin mRNA was measured, and the ratio of NOS to β-actin mRNA was calculated (NOS/β-actin). This ratio was considered representative of NOS gene expression.
Statistical analysis. All data are shown as means±standard error (SE). Statistical analyses were performed using ANOVA, followed by Scheffé’s post hoc test. Statistical significance was established at the p<0.05 level.
Results
None of the rats died during the experimental period.
Platelet counts at 4 and 8 h demonstrated similar decreases in the TF, TF+L-NIL and TF+L-NAME groups, with no significant differences between them. Similar decreases in plasma fibrinogen levels were observed in the three groups. Marked increases in plasma D-dimer and TAT levels were observed at 4 h in all three groups, and significant differences between the three groups were not observed (Table I).
Changes in plasma levels of haemostatic markers by L-NIL or L-NAME in TF-induced DIC model in rats.
The decreased platelet counts observed at 4 and 8 h in the TF group were not observed in the TF+argatroban (AR) or TF+DX9065a (DX) groups (Table II). Similarly, fibrinogen levels did not fall at 4 and 8 h in the TF+AR and TF+DX groups, as in the TF group. The marked increases in plasma D-dimer and TAT levels observed in the TF group did not occur in the TF+AR and TF+DX groups (Table II).
Changes in plasma levels of haemostatic markers by argatroban or DX-9065a in TF-induced DIC model in rats.
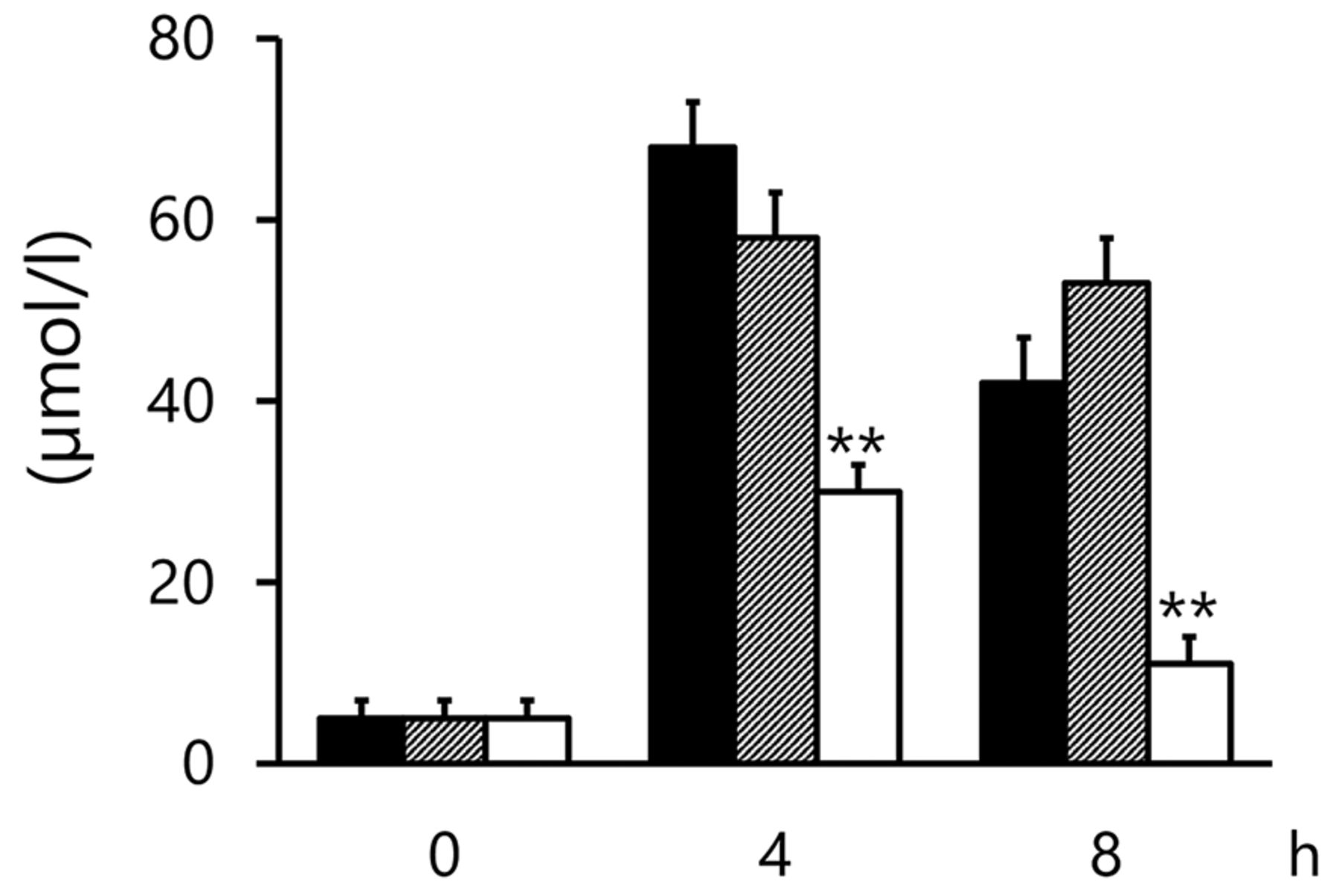
Plasma levels of NOX were obviously enhanced by TF. NOX production was significantly suppressed in the TF+L-NAME group (p<0.01 and p<0.01 at 4 and 8 h, respectively, compared with the TF group; Figure 1). However, a similar effect was not observed in the TF+L-NIL group (NS and NS at 4 and 8 h, respectively, when compared with the TF group; Figure 1). AR and DX caused marked suppression of TF-induced NOX production (p<0.01 and p<0.01 at 4 and 8 h, respectively, when TF+AR and TF+DX were individually compared with the TF group; Figure 2).

Changes in plasma NOX levels by L-NIL or L-NAME in TF-induced DIC model at 0 (pre), 4, and 8 h. Solid bars, TF group; scattered bars, TF+L-NIL group; open bar, TF+L-NAME group. Results are presented as means+S.E. **p<0.01 represents a significant difference between the TF and TF+L-NAME groups. TF: Tissue factor; L-Nil: N (6)-(iminoethyl)-lysine, iNOS inhibitor; L-NAME: L (omega)-nitro-L-arginine methylester, eNOS inhibitor.
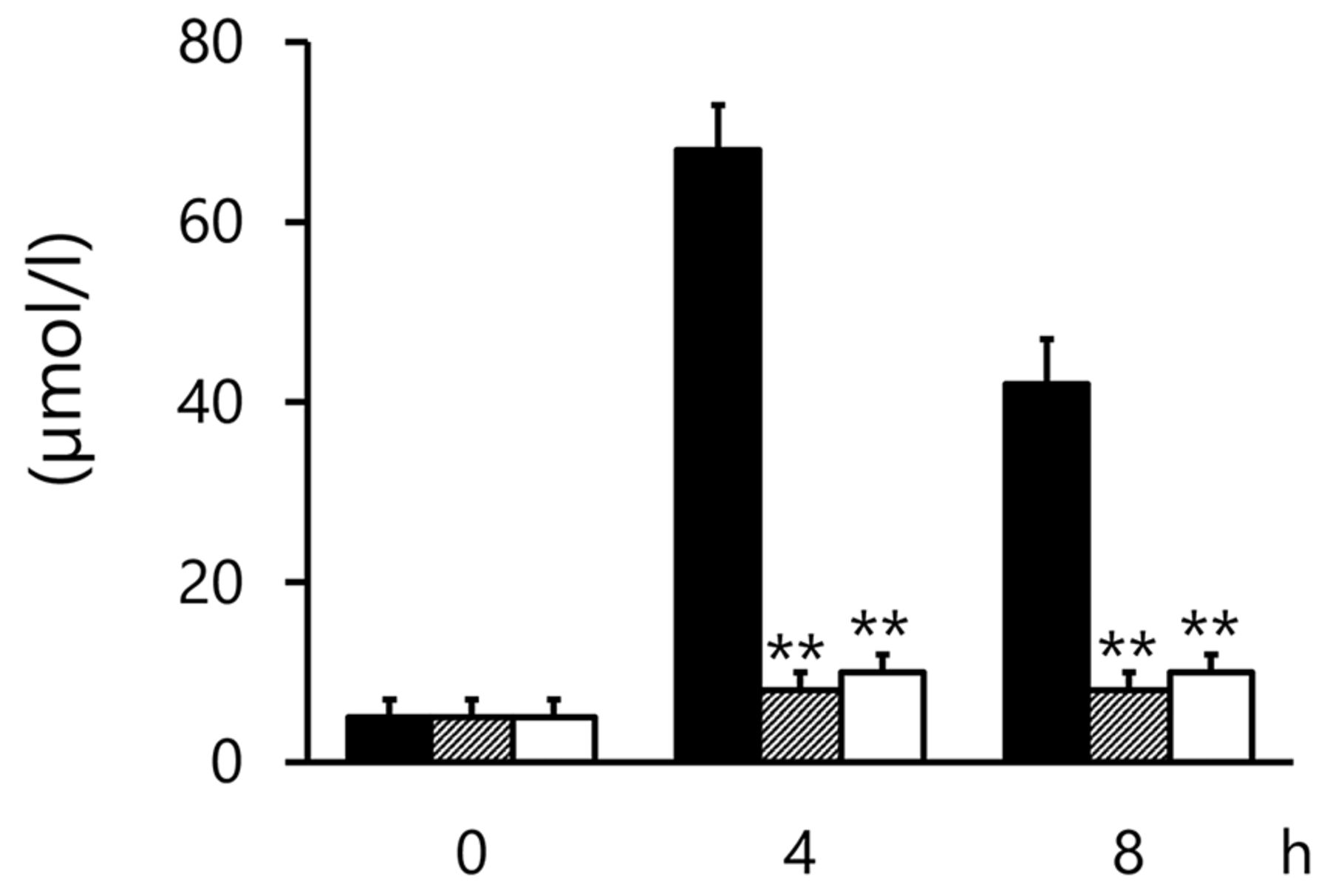
Changes in plasma NOX levels by argatroban or DX-9065a in TF-induced DIC model levels at 0 (pre), 4, and 8 h. Solid bars, TF group; scattered bars, TF+argatroban group; open bar, TF+DX-9065a group. Results are presented as means+S.E. **p<0.01 represents a significant difference between the TF and TF+argatroban groups or the TF and TF+DX-9065a groups. TF: Tissue factor.
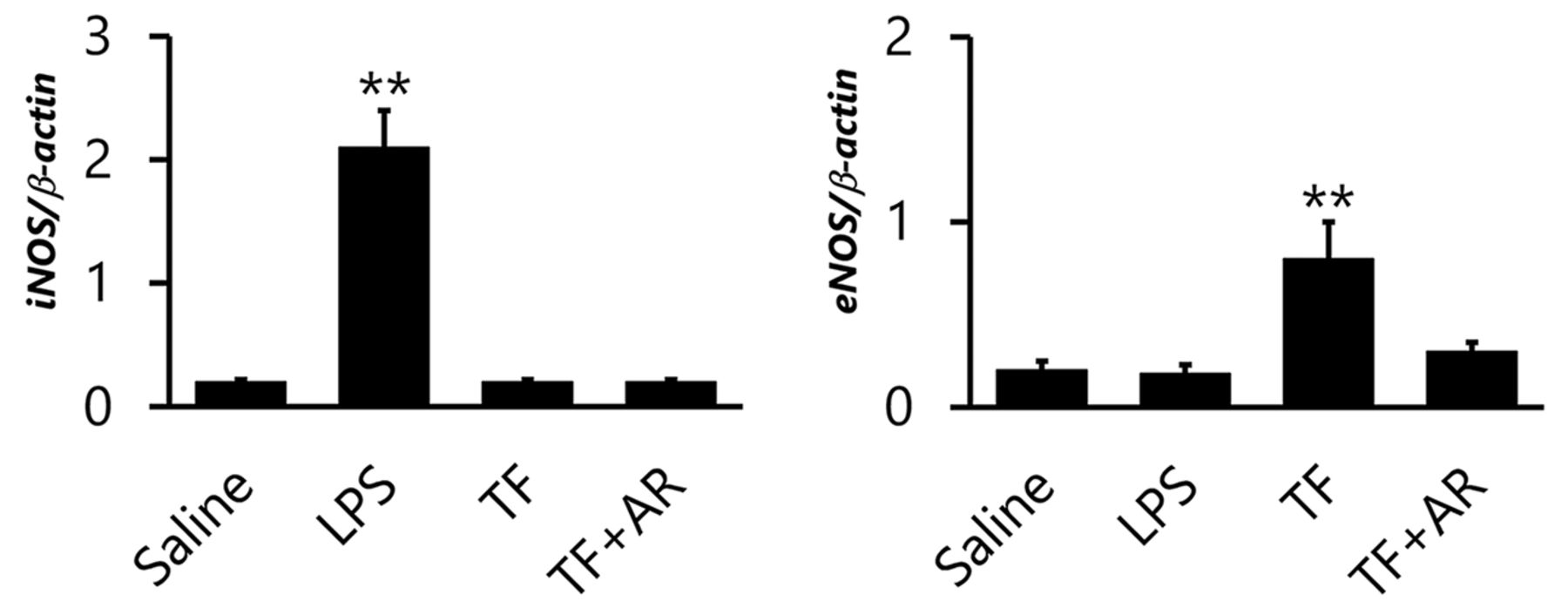
Expression of iNOS-mRNA was markedly increased in the LPS group, whereas expression of iNOS-mRNA was similar in the saline control group, the TF group, and the TF+AR group (Figure 3). On the other hand, significantly increased expression of eNOS-mRNA was observed in the TF group, compared with the control group, and marked suppression of eNOS-mRNA expression was observed upon co-administration of AR. The LPS group had similar eNOS-mRNA expression as the control group.

Relative mRNA expression of iNOS and eNOS in the lung of rats treated with saline, LPS, TF, and TF+AR. LPS: Lipopolysaccharide group; TF: tissue factor group; TF+AR: tissue factor+argatroban group. **p<0.01 represents a significant difference compared with the saline group.
Discussion
eNOS-produced NO has several important effects, such as relaxation of vascular smooth muscle (28) and suppression of platelet function (29). However, iNOS-induced NO also has a variety of effects on the vascular system and microcirculation. Especially in severe infection, iNOS produces large amounts of NO (20, 21, 30). Although several studies have demonstrated the role of NO in sepsis, its role in DIC is not well known. We previously demonstrated that iNOS-mediated NO production results in hypotension (depressed MAP), progression of hepatic and renal dysfunction, as well as deposition of microthrombi and elevated endothelin levels during LPS-induced DIC in rats (31). To the best of our knowledge, only one report has documented excess NO production in TF-induced DIC (19), and the mechanism of NO production remains unclear. Here, we attempted to clarify the factors that influence NO production during TF-induced DIC in rats, using two NOS inhibitors and two anticoagulants.
Co-administration of L-NIL or L-NAME during TF-induced DIC did not affect various hemostatic markers. In fact, an analysis of TAT levels indicated that neither L-NIL nor L-NAME altered hemostatic activation owing to DIC in the present study. Moreover, platelet counts and fibrinogen levels indicated that neither L-NIL nor L-NAME inhibited ‘consumption coagulopathy’ in the TF-induced DIC model examined in this study. Interestingly, TF-induced elevations of plasma NOX were significantly suppressed by co-administration of a relatively specific eNOS inhibitor (L-NAME), but not by a specific iNOS inhibitor (L-NIL). This contrasts our findings regarding LPS-induced DIC, in which LPS-induced elevations of plasma NOX levels were suppressed by co-administration of a specific iNOS inhibitor (31). Thus, NO production might be primarily mediated by eNOS in TF-induced DIC, and iNOS in LPS-induced DIC.
Administration of a specific thrombin inhibitor (AR), or a specific Xa inhibitor (DX), during TF-induced DIC markedly suppressed thrombin production and secondary fibrinolysis, judging from the resultant TAT and D-dimer levels. Moreover, platelet counts, and fibrinogen levels indicated that AR and DX suppressed ‘consumption coagulopathy’ in TF-induced DIC. TF-induced elevations of plasma NOX levels were almost completely suppressed by AR and DX. Judging from the effects of these anticoagulants, it is reasonable to conclude that thrombin stimulates NO production in this TF-induced DIC model.
A marked increase in iNOS-mRNA expression was observed in the LPS-induced DIC model (positive control group for iNOS expression) of this study, which agrees with previous reports (20, 21, 30) and findings regarding the effects of an iNOS inhibitor (31). On the other hand, significant iNOS-mRNA expression was not observed in the TF-induced DIC model, further supported by the finding that elevated plasma levels of NOX are not modulated by administration of an iNOS inhibitor. This is the first report to demonstrate significant enhancement of eNOS-mRNA (not iNOS-mRNA) expression in TF-induced DIC, which explains the observed effects of an eNOS inhibitor. It is also notable that eNOS-mRNA expression was almost completely suppressed by a specific thrombin inhibitor. eNOS could improve endothelial function and increase antithrombotic potential, but the presented results showed that the anti-thrombotic agent suppressed the expression of eNOS in TF-induced DIC model. This was not surprising, because the anti-thrombotic agent significantly suppressed hemostatic activation and eNOS expression was not necessary to be enhanced and remained in the levels of the control saline group.
It appears, that eNOS (not iNOS) plays an important role in the production of NO in this TF-induced DIC model. Furthermore, thrombin significantly enhances eNOS-mRNA expression in this model. In the LPS-induced DIC model, marked thrombin production was also observed, however, significant eNOS-mRNA was not. The reason for these differences is not clear, however, one hypothesis is that stimulation of eNOS-mRNA expression by thrombin is abolished by LPS, which is known to down-regulate eNOS-mRNA expression (9, 22).
It is a mysterious phenomenon that little organ dysfunction is observed in TF-induced DIC model in spite of excess thrombin generation (32). An improved microcirculation due to moderate vasodilation by NO (role of eNOS), as well as enhanced fibrinolysis, may contribute to the protection of organs in the TF-induced DIC model. However, further study is necessary to apply eNOS modulators to the therapy of DIC.
Footnotes
This article is freely accessible online.
Authors’ Contributions
Suga Y and Asakura H contributed to the experimental design, and analysis of experiments, and wrote the manuscript. Takahashi Y contributed to the experiments. Yamada S assisted with the experiments and their analysis. Morishita E contributed to drafting/editing of the manuscript and assisted with the analysis of experiments.
Conflicts of Interest
The Authors declare that they have no conflicts of interest in relation to this study.
- Received April 2, 2021.
- Revision received April 13, 2021.
- Accepted April 14, 2021.
- Copyright © 2021 International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.