


Abstract
Background/Aim: The pathway of initiation of psoriasis comprises the differentiation and infiltration of T-helper 17 (Th17) cells into the skin, characterized by the production of interleukin 17A and 17F (IL-17A/IL-17F) among other cytokines, resulting in a downstream cascade of events. Due to the lack of simplicity in psoriasis models, we aimed to develop an easily and rapidly inducible mouse model for the IL-23/IL-17 pathway with quick readouts from a straightforward lavaging process and with detectable cytokine levels. Materials and Methods: We utilized the 6-day air-pouch mouse model, injected with a combination of anti-CD3, IL-23 and IL-1β. At 24, 48 and 72 h, intra-pouch secretion of IL-17A, IL-17F and C-X-C motif chemokine ligand 1 were measured. Skin biopsies were collected and immune cell infiltration evaluated, and intra-pouch immune cells were isolated and analyzed. Results: The combination of anti-CD3, IL-23 with and without IL-1β significantly increased intra-pouch levels of IL-17A/IL-17F at 24 and 72 h, triggering a downstream production of C-X-C motif chemokine ligand 1. The cytokines were detectable even 72 h post-induction. T-cell receptor beta was down-regulated on CD4+ and CD8+ T-cells, indicating intra-pouch T-cell activation. Anti-CD3 induced CD3+ cell migration into the subcutis and the lining tissue surrounding the cavity of the air pouch, where in the latter, a similar distribution pattern of Il17a mRNA-expressing cells was also observed. However, no Th17 cell differentiation nor changes in IL-17A+ granulocytes were observed. Conclusion: The induced air-pouch mouse model induced with a cocktail of anti-CD3, IL-23 with or without IL-1β is able to mirror the IL-23/IL-17 axis of psoriasis-like inflammation characterized by immune cell infiltration and cytokine secretion.
Psoriasis is a chronic cutaneous inflammatory disease, which affects a considerable part of the global population. The most common psoriatic form is called psoriasis vulgaris, comprising 90% of the cases. Psoriatic lesions feature a well-demarcated area, usually erythematous, with silvery-white scales, typically distributed at sites of the skin, with predilection including forearms and shins, elbows and knees, scalp, peri-umbilical and peri-anal regions (1, 2).
Histologically, psoriatic skin plaques are characterized by hyperproliferation of keratinocytes (hyperplasia, acanthosis), retention of keratinocyte nuclei in stratum corneum (parakeratosis) and considerable numbers of immune cells infiltrating into the dermis and epidermis. Accumulation of neutrophils in the epidermis forms the so-called pustules of Kogoj (in the stratum spinosum) and Munro’s microabscesses (in the stratum corneum) (3). Elongation of epidermal rete ridges occurs, and newly formed blood vessels appear on the tips of dermal papillae (1).
One of the suggested pathways of the disease development, also known as the Koebner phenomenon, is initiated by a mild trauma that damages keratinocyte integrity, releasing nucleic acids into the epidermis. The fragments of the self-DNA couple with the antimicrobial peptide cathelicidin and form complexes, which have been suggested to inappropriately activate dendritic cells (4-6). The production of IL-23 by the latter promotes the differentiation of naive T-cells towards the T-helper 17 (Th17) lineage (7, 8). Th17 cells migrate into the skin, and secreted IL-23 persistently triggers the production of the cytokines IL-17A and IL-17F (9, 10). IL-17 is a potent pro-inflammatory cytokine, inducing the secretion of several chemoattractants from keratinocytes, mast cells and macrophages, including C-X-C motif chemokine ligand 1 (CXCL1), predominantly attracting neutrophils to the site of inflammation (9).
Th17 (CD4+) cells and CD8+ T-cells (Tc17) are not the only cells which secrete IL-17. A distinct subtype in mice, the skin-resident γδ T-cells, known as dendritic epidermal T-cells, can also produce IL-17 during the wound-healing process, during an infection, or even when stimulated with IL-23, IL-1β and anti-CD3 (11-13). In addition, innate immune cells, specifically mast cells and neutrophils, have been demonstrated to produce vast amounts of IL-17, released by degranulation and neutrophil extracellular traps, respectively, in patients with psoriasis (14).
The importance of the IL-23/IL-17 axis in psoriasis has been recognized, as newly developed antibodies interfering with this particular pathway have been successfully utilized for psoriasis treatment. In order to understand the disease factors and mechanisms in more detail, more simplified mechanistic mouse models mirroring a potent psoriasis-like inflammatory reaction are needed. Therefore, we aimed to induce the IL-23/IL-17 inflammation pathway by injecting a cocktail of anti-CD3, IL-23 and IL-1β in a 6-day air pouch mouse model. The choice of the air pouch model enabled us to induce a fast, localized and measurable inflammatory response (15). We hypothesized that injecting anti-CD3, IL-23 and IL-1β into the air-pouch cavity of mice would result in activation and differentiation of T-cells into the Th17 phenotype, inducing the secretion of IL-17A and IL-17F cytokines and the subsequent CXCL1 chemokine release. Although, there was no clear evidence of Th17 cell differentiation, there was a prominent response observed with intra-pouch cytokine and chemokine secretion and T-cell activation, and also a vast immune cell infiltration into the subcutis.
Materials and Methods
Animals. C57BL/6 (Charles River; Sulzfeld, Germany) female mice aged 6 to 8 weeks were housed in conventional type III cages (Techniplast, Varese, Italy) in a random distribution, at 20-23°C and 30-60% relative humidity. A common rodent pellet diet (Altromin 1413; Brogaarden, Denmark) and tap water were offered ad libitum. The permission for these animal experiments was gained from the Danish Veterinary and Food Administration, part of the Ministry of Environment and Food of Denmark (permit no. 2017-15-0201-01165).
Experimental design. Due to the model optimization, we decided to test two mice per group in each experiment, except for one group which contained three mice. Several experiments were performed, refining mainly the protocol and buffers used. Overall, the addition of 0.75% w/v carboxymethylcellulose in the injectable medium and the use of 18-gauge needle on a syringe for lavaging (see below) were among the most important milestones in model development. Sacrifice and samplings were performed daily at 24 and up to 72 h post-injection of the inflammatory triggers. In total, four experiments are presented here. An initial antibody titration study in a time-course set-up was performed, where the dose of 2.5 μg/ml of anti-CD3 was selected to be used for the remaining studies. One study was dedicated only for histological analyses (hematoxylin and eosin staining, immunohistochemistry and in situ hybridization). Two of the studies were conducted in parallel: a) Use of different combinations of inflammatory triggers at 24 h post-injection; and b) groups of mice received the full cocktail of triggers or plain medium (control) and were sacrificed at 48 and 72 h. Whenever possible, data from the groups of mice receiving same combination of triggers and having same endpoints were pooled together to increase the sample size, while allowing the statistical analysis to be subject to inter-study data heterogeneity. More information about the components of the injected medium is mentioned in the Medium and inflammatory triggers section.
Air pouch establishment. We used a modified version of the air-pouch mouse model from Sin et al. (16), first described by Edwards et al. (17). On day 0 and 3, mice were injected subcutaneously in the dorsal back area with 2.5 ml of sterile air passed through a sterile syringe filter (0.2 μm Millipore filter; Merck, Darmstadt, Germany). Prior to the injection the area was shaved and disinfected with 70% ethanol. The injections were then made with a 27-gauge needle (BD Microlance™ 3; BD Biosciences, Franklin Lakes, NJ, USA) inserted at a point between the shoulder blades, with the cranial tip of the needle pointing towards the tail of the mouse. In addition, two fingers holding onto the sides of the mouse facilitated the procedure by preventing the air from diffusing around the body and cranial area of the mouse. The pouch was situated approximately 1 cm from the caudal aspect of the head of the mouse. The injection site was squeezed with forceps for 10-15 s after withdrawal of the needle. The air pouch was allowed to establish a cavity for 6 days. On day 6, 1 ml of medium with or without the inflammatory triggers, as described below, was injected.
Medium and inflammatory triggers. The cocktail of the inflammatory triggers, injected once 6-days after the establishment of the air-pouch cavity, contained 0.25 μg/ml to 5 μg/ml monoclonal Armenian hamster anti-mouse anti-CD3s (clone 145-2C11; Biolegend, Koblenz, Germany) with/without 500 ng/ml recombinant mouse IL-23 (R&D Systems, Minneapolis, MI, USA) with/without 0 ng/ml recombinant mouse IL-1β (Biolegend) mixed in 1 ml of Iscove’s modified Dulbecco’s medium (IMDM; Gibco-Thermo Fisher Scientific, Waltham, MA, USA) supplemented with a sterile, moderately viscous solution of 0.75% w/v carboxymethylcellulose (CMC, Sigma-Aldrich; Saint Louis, MO, USA) diluted in 0.45% v/v NaCl. The control groups were injected with 1 ml IMDM (0.75% w/v CMC). Non-viscous IMDM was used only in histological studies.
Termination and lavaging. Animals were euthanized by cervical dislocation under isoflurane anesthesia, at 24, 48 and 72 h post-injection of inflammatory triggers. The pouch of each mouse was shaved with an electric razor prior to sampling. The air-pouch cavities of the mice were flushed with 0.5 ml phosphate-buffered saline containing 10 mM EDTA (room temperature) to mildly detach cells from the cavity and prevent cellular clumps, with an 18-gauge needle (BD Microlance™ 3; BD Biosciences) on a 1 ml syringe, avoiding any damage to the pouch while massaging and pipetting. The procedure was repeated again with 1 ml of phosphate-buffered saline (10 mM EDTA) to retrieve as many immune cells as possible. The lavage fluids of 0.5 ml and 1 ml were transferred into two separate Eppendorf tubes and centrifuged at 300×g for 10 min (room temperature). The supernatants of the 0.5 ml volumes were transferred to a new tube and stored at −20°C for later analysis with enzyme-linked immunosorbent assay (ELISA), while the 1 ml supernatant from the second wash was discarded. Cell pellets from the same mouse were pooled by re-suspending in 250 μl IMDM, in total, and processed for flow cytometric analysis.
Histology. Skin biopsies of 8 mm in diameter were collected with disposable biopsy punches (Integra™; Miltex™, York, PA, USA) from the center of each pouch. Tissue samples were immersed in 10% formalin (3.7% formaldehyde) (Sigma-Aldrichi) for 24 h, processed with Excelsior™ A/S Tissue Processor (Thermo Fisher Scientific) and embedded in paraffin. Hematoxylin (Fagron Nordic A/S, Copenhagen, Denmark) and eosin (Carl Roth GmbH, Karlsruhe, Germany) staining was performed on 3-4 μm deparaffinized microtomed sections. Slides were scanned with NanoZoomer (2.0-HT) (Hamamatsu Photonics, Hamamatsu, Japan), images were extracted through its viewing platform, NDP.View.v2 (Hamamatsu Photonics) based on the default image settings, and cellular infiltration as well as vasodilation were evaluated in skin and lining tissue surrounding the air pouch cavity.
Immunohistochemistry (IHC). Prior to staining, tissues slides were deparaffinized in BOND buffer at 60°C overnight. CD3+ cells were stained using a rabbit monoclonal antibody to CD3 (clone SP7, dilution 1:200; Sigma-Aldrich). A secondary goat anti-rabbit IgG, alkaline phosphatase-conjugated polyclonal antibody (Immunologic, Palo Alto, CA, USA) was used in reaction with Fast Red chromogen (BOND polymer refine red detection; Leica Biosystems, Wetzlar, Germany) for staining the antibody-bound cells. Slides were scanned and images were collected as mentioned above but with linear γ correction (γ=1) and 110% contrast. The infiltration of positively stained CD3+ cells (red colored) was evaluated in skin and lining tissue in random frames.
In situ hybridization (ISH). ISH for Il17a mRNA was performed on formalin-fixed paraffin-embedded tissue using RNAscope 2.5 Leica Assay Reagent Kit RED (Leica Biosystems; Wetzlar, Germany) according to the manufacturer’s instructions (Advanced Cell Diagnostics, Inc., Hayward, CA, USA). Hybridization was performed using ACD target probes (MmIl17a, positive control Mm PPIB and negative control Hs DapB).
All of the sections were blinded before description. All counts were performed by the same individual via visual inspections. In accordance with a recommendation from ACD, a cell profile was defined as Il17a mRNA-positive if it had at least one red spot visible at 20× magnification. A minimum of 30 randomly selected counting frames per biopsy were evaluated.
ELISA. Thawed 0.5 ml supernatants from the first lavage fluid retrieved from the air-pouch were used for cytokine and chemokine quantification. Murine IL-17A and CXCL1 were quantified by the respective Quantikine ELISA kits and mouse IL-17F cytokine with DuoSet ELISA kit (both R&D Systems), according to the manufacturer’s instructions. The recoveries of standard concentrations were 90-110% for IL-17A and 80-120% for IL-17F and CXCL1. The threshold of R2 was set at 0.995. The lower limit of quantifications of the kits were 11, 23 and 16 pg/ml for IL-17A, IL-17F and CXCL1, respectively. The upper levels of quantification were 700, 1,500 and 1,000 pg/ml, respectively. Dilutions of some samples were applied for IL-17F ELISA analysis, where the quantification limit is mentioned as twice the upper limit, i.e. 3,000 pg/ml. Thus, the quantification of the samples remained within the limits, except for two samples, measured for IL-17A, which were slightly above the limit, but acceptable.
Flow cytometry. Cells were counted by NucleoCounter NC-3000, using Vial-Cassette™ (Chemometec; Allerod, Denmark) and 106 cells per well were plated in a 96-well plate. Cells were stimulated for 5 h with phorbol 12-myristate 13-acetate (PMA) and ionomycin in the presence of protein transport inhibitors to maximize intracellular cytokine expression and subsequently their detection (Cell Stimulation Cocktail plus protein transport inhibitors 500×; eBioscience, San Diego, CA, USA). Fc blockers (anti-CD16/CD32; BD Biosciences) were used in order to minimize any non-specific binding, prior to staining. Cells were resuspended in 20 μl of Brilliant Stain buffer (BD Biosciences) and stained with fluorescein-conjugated anti-CD45, allophycocyanin -Cy7-conjugated anti-GR1(Ly6C/Ly6G), anti-CD4/BV711, anti-CD8α/BV650 and anti-T-cell receptor beta (TCRβ)/BV786 (all BD Biosciences). Cell were fixed and permeabilized with BD Cytofix/Cytoperm™ kit (BD Biosciences) according to the manufacturer’s instructions and stained with anti-IL-17A/allophycocyanin (17B7, eBioscience; San Diego, California). Compensation of fluorophores was performed with BD CompBeads™ (BD Biosciences). All the antibodies were previously titrated and dilutions with optimal Separation Index were used in the studies. Stained cells were run in LSR II flow cytometer (BD Biosciences), using BD FACSDiva™ (BD Biosciences), and analysed in FlowJo™ VX (LCC, Ashland, OR, USA). The down-regulation of TCRβ was measured through geometric mean fluorescent intensity (gMFI). Flow cytometry raw files are publicly available at http://flowrepository.org (ID: FR-FCM-Z3YB and FR-FCM-Z3ZE).
Statistical analysis. For data analysis and graph designing, GraphPad PRISM v.6.01 (GraphPad Software Inc., La Jolla, CA, USA) was used. One-way analysis of variance or unpaired t-test was carried out to determine the statistical significance of differences between groups. Values have been presented as the mean with the standard error of the mean. The threshold of the p-value was set at 0.05 for statistically significant results. Post-hoc power analysis was performed with G*Power3.1 software (18). The significance level for the post-hoc test was set at α=0.05 and the observed power was 1−β>0.95 (β: error probability) for each statistical analysis with a significant difference (analysis of variance ANOVA or t-test), confirming an adequate sample size for the analysis. In general, inter-study variations were observed in some cases, but there were not any outliers identified by ROUT method (for Q=5%). Raw data and analyses are available upon request.
Results
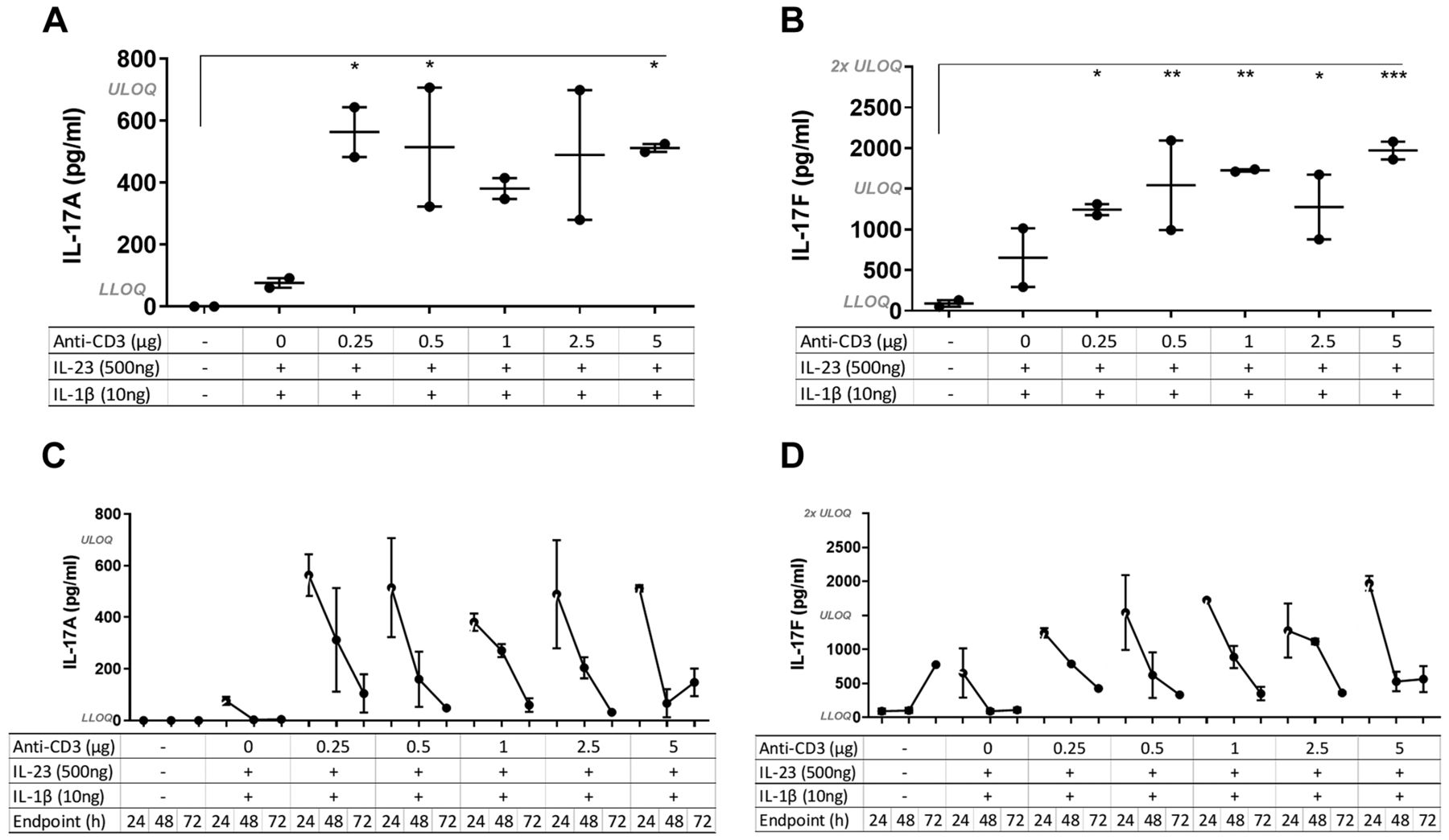
The effect of different concentrations of anti-CD3 on the inflammatory response. To determine the optimal concentration of anti-CD3 to induce an inflammatory response but not deplete the T-cells, an antibody titration study was conducted and mice were sacrificed at 24 h after administration of anti-CD3. IL-23 and IL-1β dose remained the same for all the groups (500 ng and 10 ng per mouse, respectively), except the control which received plain IMDM (0.75% w/v CMC). Even at the lowest dose of anti-CD3 (0.25 μg), the intra-pouch levels of IL-17A and IL-17F measured in the lavage fluid were significantly increased compared to control mice and those receiving only IL-23 and IL-1β (no anti-CD3) (Figure 1A and B). Furthermore, cytokine secretion was reduced throughout the time course of the study (Figure 1C and D); multiple ANOVA test was performed for all of the groups and only the significant results at 24 h are presented in Figure 1A and B.
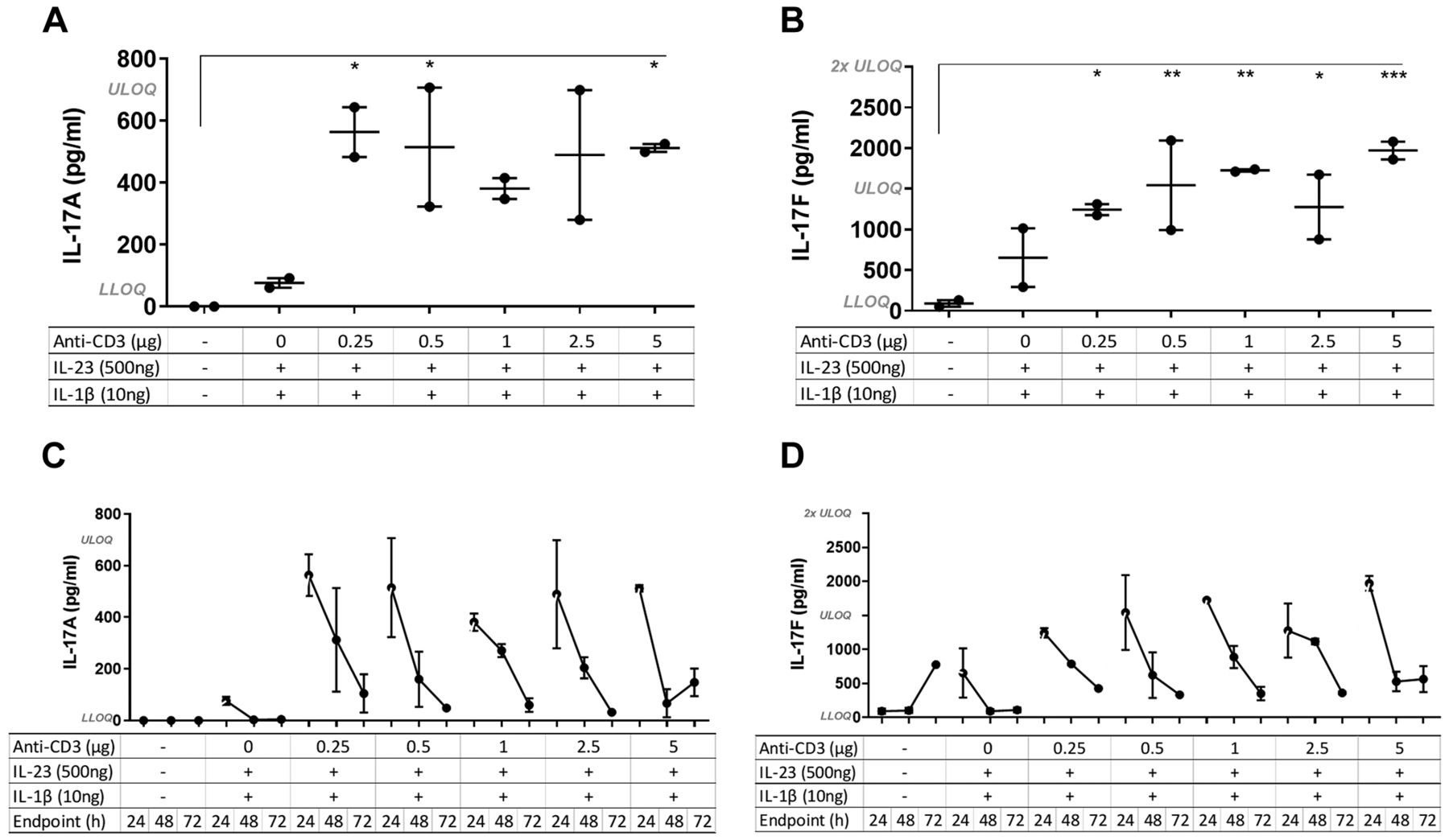
Effect of different concentrations of anti-CD3 on interleukin-17A (IL-17A) and IL-17F secretion. IL-17A and IL-17F were quantified in air-pouch lavage fluid from mice treated with Iscove’s modified Dulbecco’s medium (0.75% w/v carboxymethylcellulose) (control), or different concentrations of anti-CD3 given in a cocktail together with 500 ng/ml IL-23 and 10 ng/ml of IL-1β. Mice were sacrificed at 24, 48 or 72 h after injection of the cocktail (N=2 per group). A and B: Multiple analysis of variance was conducted for all of the time-course groups at 24-h. IL-17A and IL-17F were significantly increased at 24 h in groups treated with anti-CD3 even at the lowest concentration compared to control mice. The mean±SEM are indicated. Significantly different from control mice at: *p<0.05, **p<0.01, ***p<0.001. C and D: The levels of IL-17A and IL-17F declined over time for each group. LLOQ: Lower limit of quantification: IL-17A: 11 pg/ml; IL-17F: 23 pg/ml. ULOQ: Upper limit of quantification: IL-17A: 700 pg/ml; IL-17F: 1,500 pg/ml.
Combination of anti-CD3 and IL-23 increased intra-pouch production of IL-17 and CXCL1. To investigate the effect of each of the components in the inflammatory cocktail on the inflammatory response, different combinations of anti-CD3 and the two cytokines were tested. Anti-CD3 and IL-23 resulted in a statistically significant increase of both IL-17A and IL-17F compared to the control group (Figure 2A and B, respectively). When the mice were treated with IL-23 alone or in the presence of IL-1β, there was a significant increase observed in IL-17F production compared to the control groups. However, under these circumstances, IL-17A was only detected in trace amounts. In addition, anti-CD3 administration alone resulted in a statistically significant increase in production of IL-17F but not of IL-17A, although its level was still elevated compared to the control mice and mice treated with IL-23 (Figure 2A and B). Thus, the inclusion of anti-CD3 in the inflammatory cocktail significantly amplified the production of both IL-17 cytokines in the air-pouch cavity.

Effect of different combinations of the inflammatory triggers on intra-pouch production of interleukin-17A (IL-17A) (A), IL-17F (B) and C-X-C motif chemokine ligand 1 (CXCL1) (C). Intra-pouch secretion in mice treated with different combinations of the inflammatory triggers in mice sacrificed 48 h after the injection of the inflammatory triggers (N=2 per group). Control mice received Iscove’s modified Dulbecco’s medium (IMDM) (0.75% w/v carboxymethylcellulose). IL-17A and IL-17F production was significantly increased in anti-CD3, IL-23 treated groups and CXCL1 was increased in all anti-CD3-treated mice. The mean±SEM are indicated. Significantly different from control mice at: *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001. LLOQ: Lower limit of quantification: IL-17A: 11 pg/ml; IL-17F: 23 pg/ml; CXCL1: 16 pg/ml. ULOQ: Upper limit of quantification: IL-17A: 700 pg/ml; IL-17F: 1,500 pg/ml; CXCL1: 1,000 pg/ml.
Moreover, a downstream effect of the inflammatory triggers was observed, as intra-pouch secretion of CXCL1 was detected. The quantification of CXCL1 varied between the groups but was increased in all anti-CD3-treated mice (Figure 2C) and this was related to IL-17A and IL-17F secretion.
Time-course study of the inflammatory response in the air-pouch cavity. To determine the longevity of the inflammatory reaction in the air-pouch model, a 3-day time-course study was performed. Mice were injected with 2.5 μg/ml anti-CD3, 500 ng/ml IL-23 and 10 ng/ml IL-1β and sacrificed at 24, 48 or 72 h post induction.
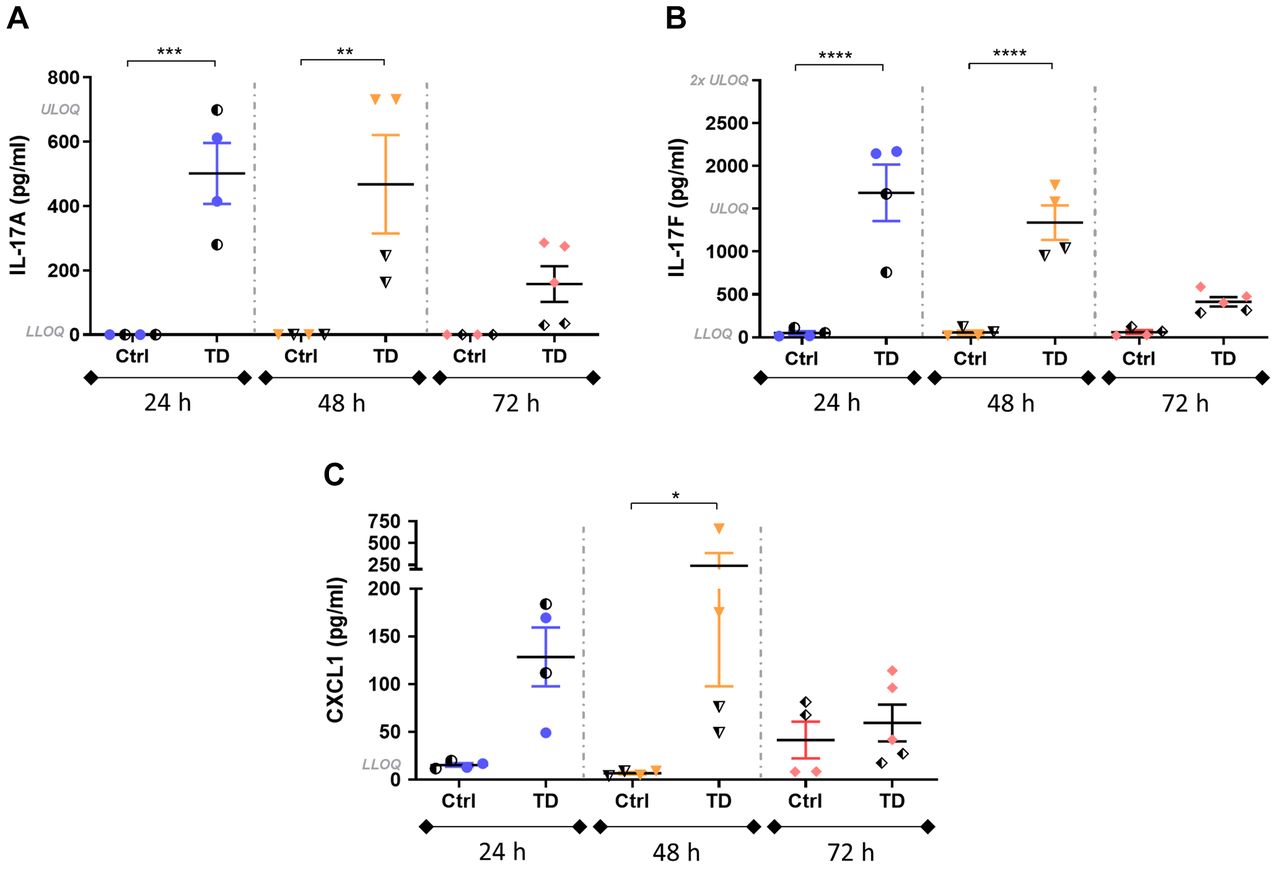
Intra-pouch secretion of IL-17A and IL-17F was significantly increased in the induced mice, particularly at 24 and 48 h post induction compared to the respective control groups (Figure 3A and B). The highest peak of both IL-17 cytokines was observed at 24 h, with a tendency to decrease thereafter. At 72 h post induction, the differences between control and induced groups were non-significant but still notable. The detection of IL-17 cytokines in control groups was quite low at all time points and in some cases was below the lower limit of quantification.
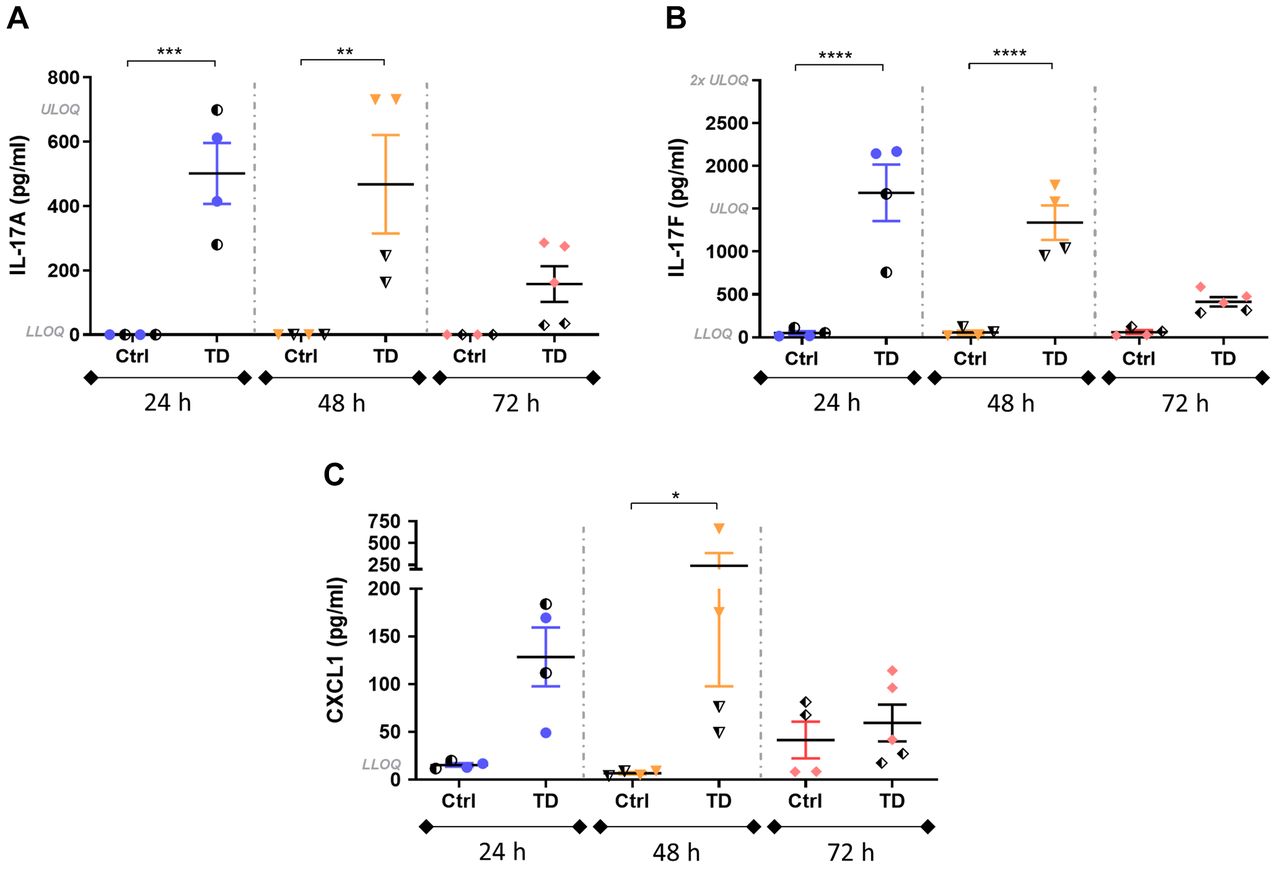
Intra-pouch secretion of interleukin-17A (IL-17A), IL-17F and C-X-C motif chemokine ligand 1 (CXCL1) at 24-, 48- and 72-h post-induction in control mice (Iscove’s modified Dulbecco’s medium + 0.75% w/v carboxymethylcellulose) and mice treated with 2.5 µg/ml anti-CD3, 500 ng/ml IL-23 and 10 ng/ml IL-1β (TD) then sacrificed 24, 48 and 72 h later (N=4-5 per group). IL-17A and IL-17F were significantly increased at 24 and 48h post-induction, while a noticeable increase of CXCL1 was observed. Half-solid symbols indicate samples from mice treated in the same way within the respective groups, collected from the antibody titration study. The mean±SEM are indicated. Significantly different from control mice at: *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001. LLOQ: Lower limit of quantification: IL-17A: 11 pg/ml; IL-17F: 23 pg/ml; CXCL1: 16 pg/ml. ULOQ: Upper limit of quantification: IL-17A: 700 pg/ml; IL-17F: 1,500 pg/ml; CXCL1: 1,000 pg/ml.
Intra-pouch production of CXCL1 was also increased in treated mice at 24 and 48 h, decreasing at 72 h (Figure 3C). Although differences between control and treated mice at 24 and 48 h were notable, they were only significant at 48 h.
To determine whether T-cells were activated in the air-pouch cavity, we performed flow cytometric analysis of the cells from the lavage fluid in the air pouch at different time points after the injection of inflammatory triggers. The level of TCRβ was found to be down-regulated, indicating a potential T-cell activation in the induced mice (Figure 4). Specifically, at 72 h the mean gMFI was significantly decreased compared to the control group (Figure 4).
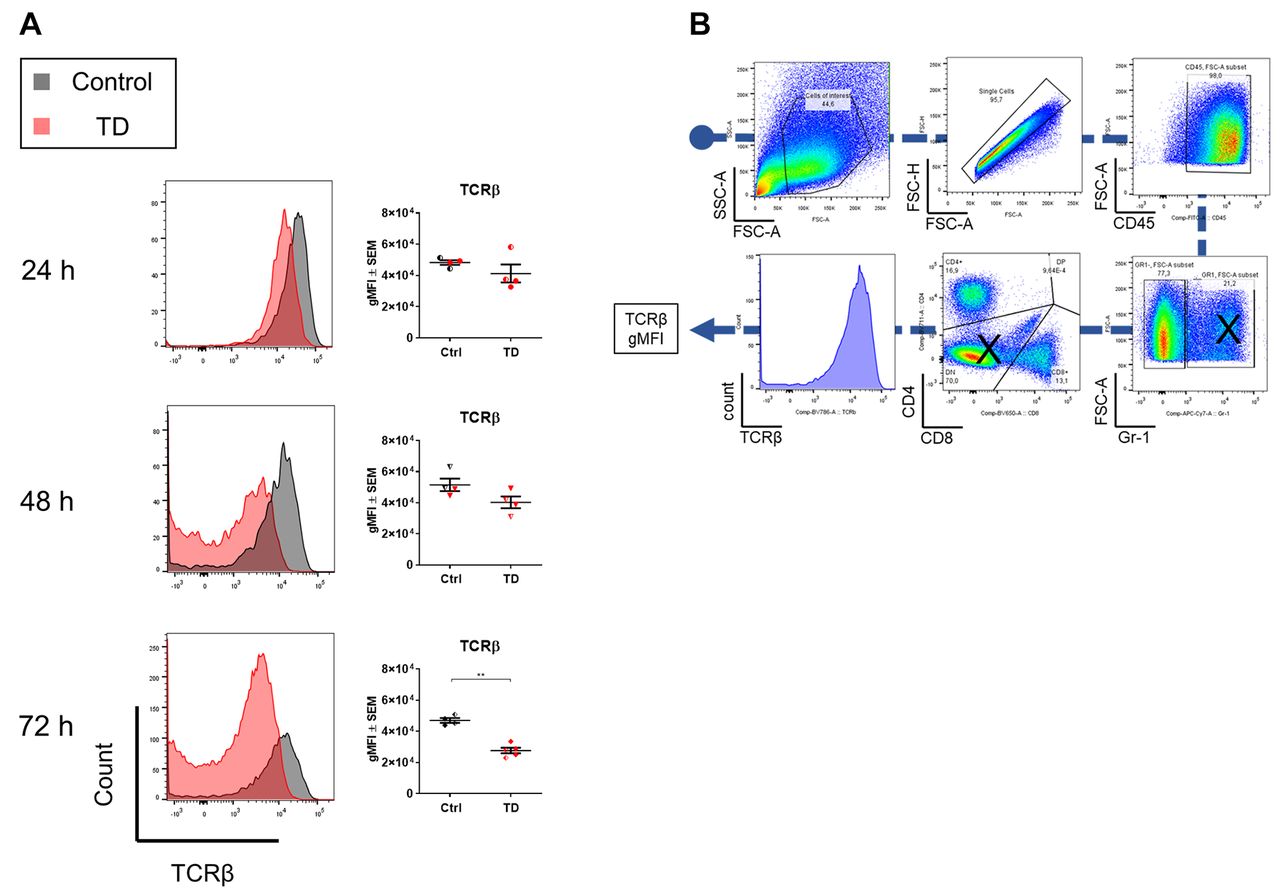
Evaluation of T-cell receptor beta (TCRβ) down-regulation in the air-pouch cavity at 24, 48 and 72 h post treatment. A: Cells from lavage fluid were characterized by flow cytometry. Visualization of TCRβ expression with overlaid histograms and TCRβ geometric mean fluorescent intensity (gMFI), calculated by FlowJo, of control (Iscove’s modified Dulbecco’s medium + 0.75% w/v carboxymethylcellulose) and treated mice (TD) [2.5 µg/ml anti-CD3, 500 ng/ml interleukin-23 (IL-23) and 10 ng/ml IL-1β], sacrificed at 24, 48 or 72 h (N=4-5 per group). Mice treated with anti-CD3, IL-23 and IL-1β showed down-regulation of TCRβ on the surface of T-cells, indicating T-cell activation. Half-solid symbols indicate samples from mice treated in the same way within the respective groups, collected from the antibody titration study. **Significantly different at: p<0.01. B: Gating strategy: GR1+ cells and CD4−CD8− cells were excluded to minimize any false-positive signals.
The presence of anti-CD3 triggered increased vasodilation and CD3+ cell infiltration in the subcutis. We characterized the air pouch by histology and the impact of the anti-CD3 administration was examined in histological samples of the air pouch. Markedly increased vasodilation and immune cell infiltration, mainly in the subcutis, was observed in the anti-CD3-treated group (Figure 5). In addition, the extravasated immune cells in the subcutis were in proximity to the lining tissue which surrounded the air pouch cavity below the cutaneous muscle layer. Intriguingly, a large number of the infiltrating immune cells positively stained for the CD3 marker in immunohistochemistry, indicating the presence of T-cells or γδ T-cells, or even invariant natural killer T-cells due to the inflammatory reaction caused by the cocktail of inflammatory triggers (Figure 6).
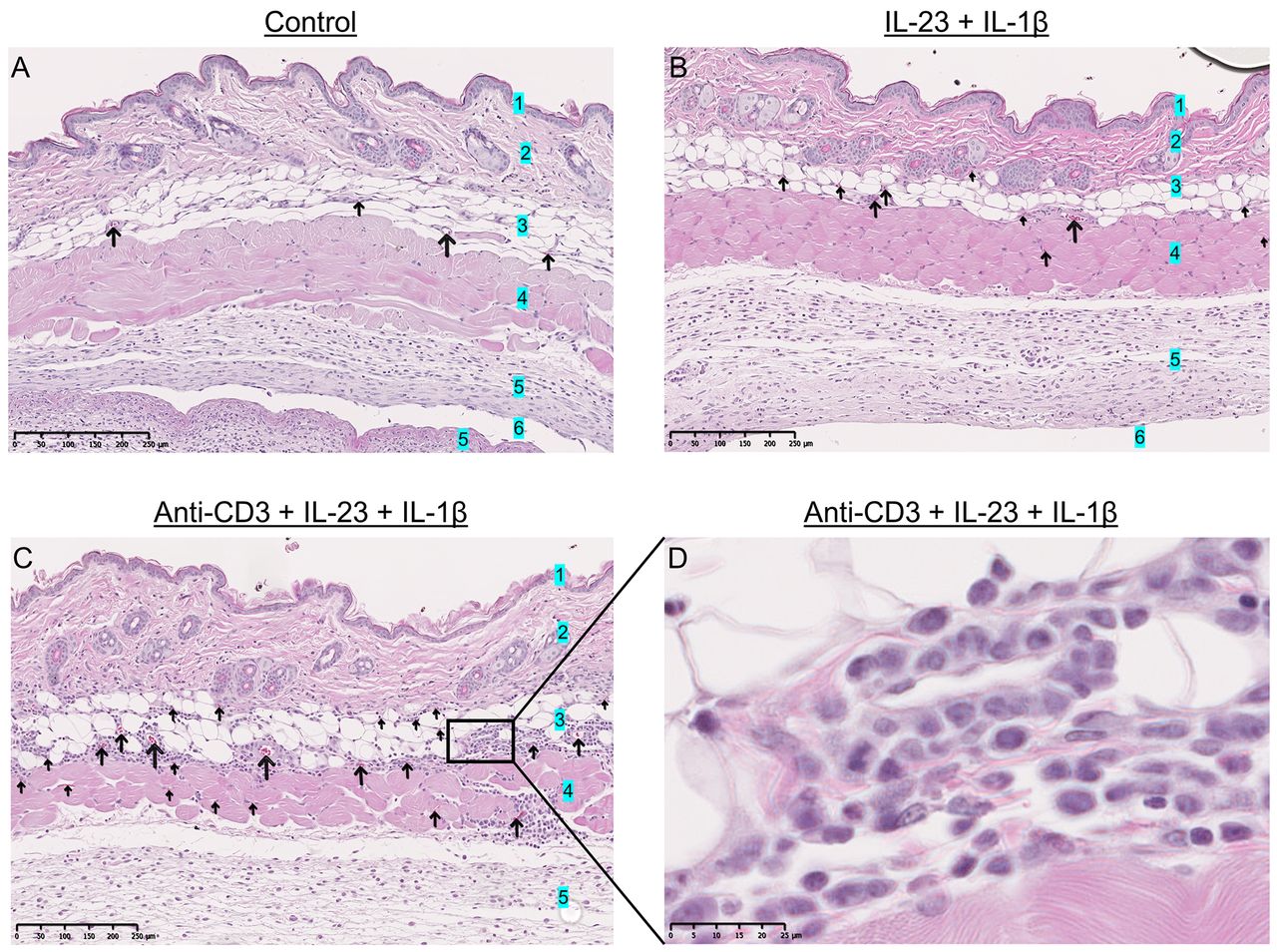
The effect of anti-CD3 as demonstrated in histological samples. Vasodilation and immune cell infiltration assessed by hematoxylin and eosin staining of back skin samples containing the air pouch cavity in control mice (A) (Iscove’s modified Dulbecco’s medium) and mice treated with 500 ng/ml interleukin-23 (IL-23) and 10 ng/ml IL-1β (B), 2.5 µg/ml anti-CD3 and 500 ng/ml IL-23 with 10 ng/ml IL-1β (C and D). Mice treated with anti-CD3, IL-23 and IL-1β exhibited increased vasodilation and immune cell infiltration, mainly in the subcutis. Arrows: Dilated vessels. 1: Epidermis, 2: dermis, 3: subcutis, 4: subcutaneous muscle layer, 5: lining tissue, 6: air-pouch cavity.
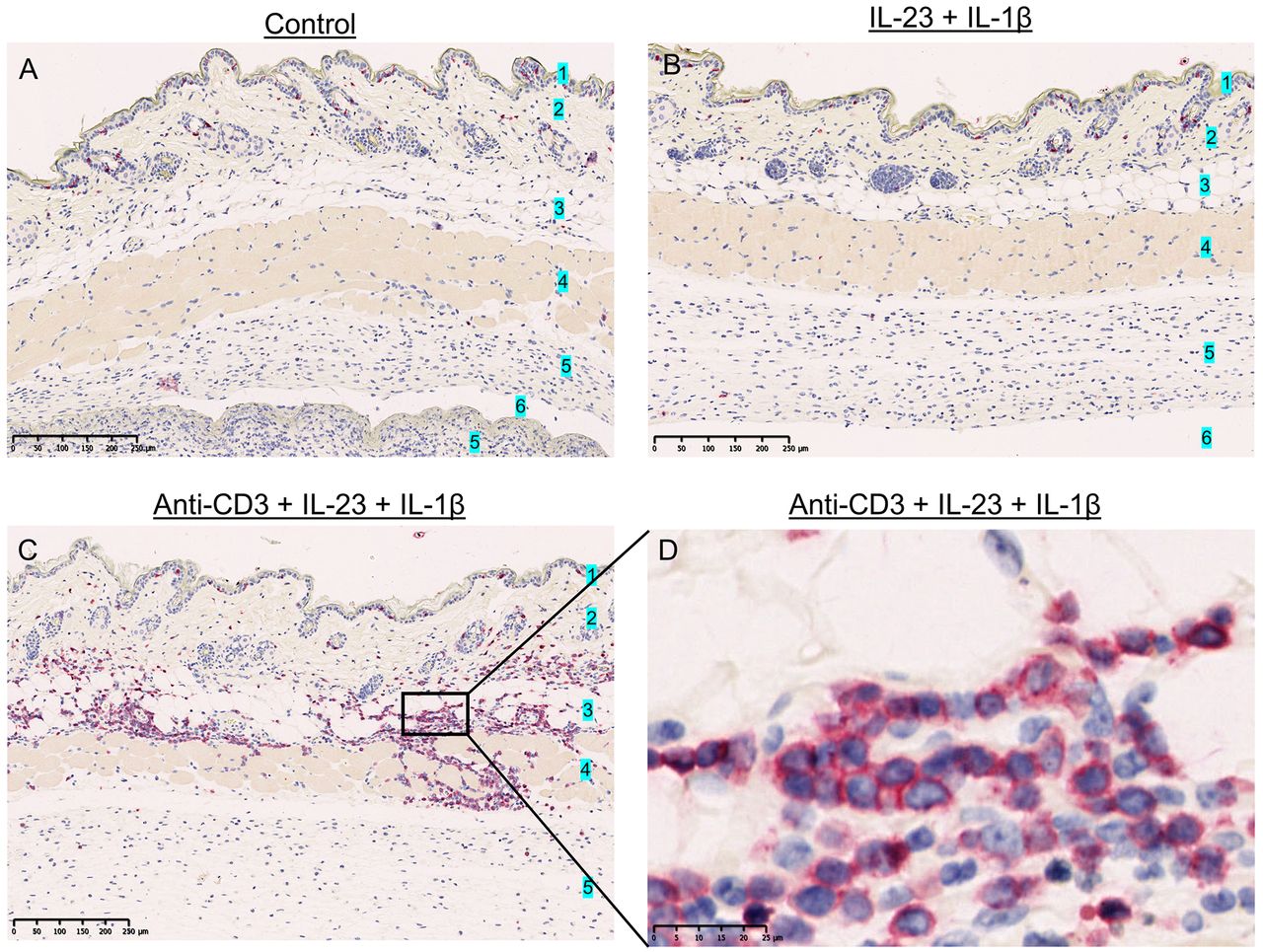
CD3+ cell infiltration in histological samples from air-pouch skin samples. CD3 immunohistochemical staining of skin samples containing the air-pouch cavity in control mice (Iscove’s modified Dulbecco’s medium) (A) and mice treated with 500 ng/ml interleukin-23 (IL-23) and 10 ng/ml IL-1β (B), and 2.5 µg/ml anti-CD3, 500 ng/ml IL-23, 10 ng/ml IL-1β (C and D). Mice treated with anti-CD3, IL-23 and IL-1β exhibited increased CD3+ cell infiltration, mainly in the subcutis. CD3+ cells stained with red color. 1: Epidermis, 2: dermis, 3: subcutis, 4: subcutaneous muscle layer, 5: lining tissue, 6: air-pouch cavity.
Expression of Il17a in the air pouch by mRNA ISH. We found that administration of anti-CD3 was associated with a notable increase in the number of cells exhibiting Il17a mRNA expression within the lining tissue surrounding the air-pouch cavity. This was seen at both 48 and 72 h after administration of anti-CD3, IL-23 and IL-1β (Figure 7). Expression of Il17a mRNA was also present in control groups at 48 and 72 h but, interestingly, we observed a difference in the expression pattern of the treated groups compared to the control groups. In addition to the small dots of mRNA expression in most of the cells (mostly at 72 h control and all of the treated mice), some single cells showed an expression pattern with a large amount of positive staining only in the treated groups, present at 24, 48 and 72 h (Figure 7). This might suggest that some randomly distributed single cells in the lining tissue had a more pronounced expression of Il17a in the treated groups compared to control groups.
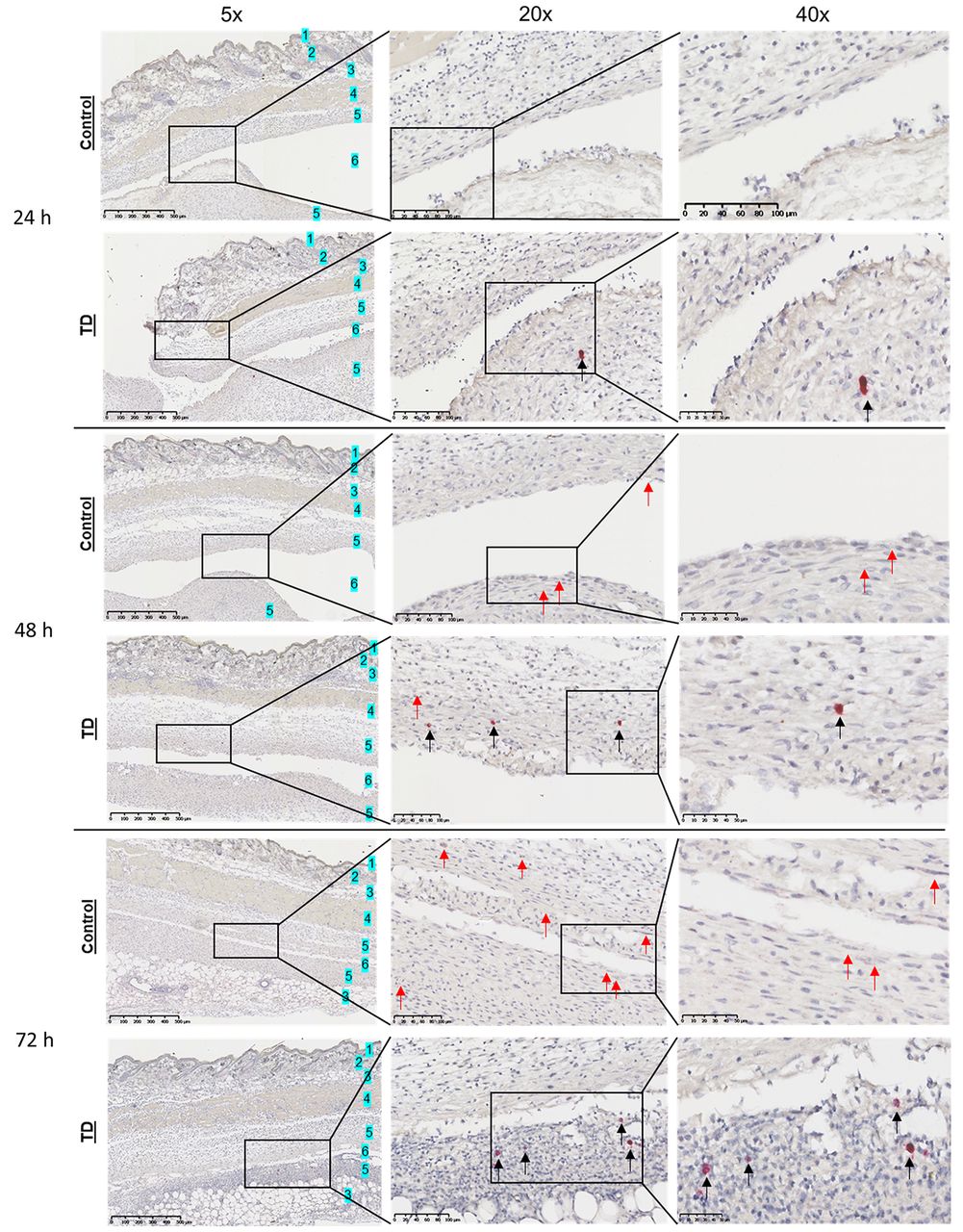
Increased interleukin-17A (Il17a) mRNA expression in the treated (TD) groups at 24, 48 and 72 h after induction. Representative images are shown for Il17a RNAScope in situ hybridization from the air pouch skin puncture at 24, 48 and 72 h for the control (Iscove’s modified Dulbecco’s medium) and groups induced with 2.5 µg/ml anti-CD3, 500 ng/ml IL-23 and 10 ng/ml IL-1β. Red arrows indicate mRNA-positive cells in each image. Black arrows indicate the presence of cells with a more pronounced expression of Il17a. Overall, small positive dots are observed in induced groups and 72 h control mice. Scale bars: 500 µm (5×), 100 µm (20×), 50 µm (40×). 1: Epidermis, 2: dermis, 3: subcutis, 4: subcutaneous muscle layer, 5: lining tissue, 6: air pouch cavity.
Presence of IL-17A producing cells in the air pouch lavage fluid. Apart from T-cell activation via down-regulation of TCRβ and the histological analyses, we also evaluated the infiltration of IL-17A producing cells or the differentiation of T-cells to Th17 in the air pouch lavage fluid by intracellular staining via flow cytometry. Cell suspensions were collected through lavaging the air pouch, washed and stimulated with PMA/ionomycin in presence of protein transport inhibitors for 5 h to induce a detectable expression of intracellular IL-17A.
We observed an increased infiltration of immune cells in the cavity, but no considerable difference between control and induced groups (Figure 8A and B). Both GR1+CD45+ (granulocytes, monocytes and macrophages) and other immune cells among the T-cells (GR1–CD45+) seemed to express IL-17A after stimulation with PMA/ionomycin. The Gr1–CD45+ cells were further evaluated for differentiation of Th17 (CD4) and Tc17 cells (CD8). However, there was no significant difference between the groups (Figure 8C-F).

Intracellular expression of interleukin-17A (IL-17A) in GR1+CD45+ granulocytes/monocytes and GR1-CD45+ leukocytes among T-cells from control (Iscove’s modified Dulbecco’s medium) and induced mice (TD: 2.5 µg/ml anti-CD3, 500 ng/ml IL-23, 10 ng/ml IL-1β). A and B: There were no noticeable differences regarding neutrophil infiltration (GR1+CD45+) in the air pouch cavity of control and induced mice at 24-72 h. C-E: Intracellular IL-17A expression in GR1+ and GR1-populations showed no significant change, while there was a noticeable inter-study variation in some of the groups. F: Potential memory T-helper 17 (CD4+IL-17A+) and Tc17 (CD8+IL-17A+) cells were observed both in control and induced mice although in very small fractions.
Discussion
IL-17 and CXCL1 are some of the well-studied biomarkers in human psoriasis (19). Thus, the air-pouch model with optimized induction of inflammation and the ease of quantifying relevant biomarkers by ELISA comprises a useful animal model in drug discovery.
In the experiments investigating the importance of each component of the inflammation-inducing cocktail, it was very clear that anti-CD3 played a crucial role (Figure 2), resulting in high levels of IL-17 in the preformed air-pouch cavity. Thus, induced CD3+ cells, such as T-cells and γδ T-cells or even the less abundant invariant natural killer T-cells, comprise the major cell types most likely to be involved in the production of these cytokines. Anti-CD3 also led to a potent induction of inflammatory hallmarks in the histological sections of the air pouch, where anti-CD3-treated mice had a more pronounced vasodilation and influx of immune cells compared to the mice treated with vehicle or cytokines only (Figure 5). Many of the infiltrating mononuclear cells stained positively for the CD3 marker (Figure 6), which indicates that anti-CD3 indeed activated T-cells, confirmed by the down-regulation of TCRαβ receptor by flow cytometry (Figure 4), resulting in a pronounced T-cell influx into the subcutis, with a tendency to move towards the lining tissue of the air-pouch cavity.
Albeit the accumulated CD3+ cells in the subcutis were found to be Il17a-negative in ISH staining, the low frequency of CD3+ cells located in or migrating to the lining tissue mirrors the low number of more pronounced Il17a-expressing cells found in this area, which could potentially comprise the major source of IL-17 secreted in the air-pouch cavity (Figure 7). However, an overlay of the two stainings is needed in order to draw any firm conclusions on this.
The time-course studies demonstrated an optimal window of inflammation at 24 to 48 h post injection of the inflammatory cocktail (Figure 3) and this seems rather quick if the measured biomarkers were produced by de novo differentiated Th17 cells. In vitro, the combination of anti-CD3, IL-23 and IL-1β can lead to T-cell differentiation into the Th17 phenotype within a period of 3 to 6 days (20, 21). Considering also the lack of a significant number of Th17 cells in flow cytometry (Figure 8F), we excluded newly differentiated Th17 cells as being the major source of these cytokines, while memory Th17 and γδ T-cells still comprise a candidate source.
We speculate that in the air-pouch model there are only few Th17 cells, which after stimulation with anti-CD3 and IL-23 produce vast amounts of IL-17. These cells might be memory rather than newly differentiated cells. This may also explain why we did not detect large fractions of Th17 cells in the lavage fluid (Figure 8F). This hypothesis is further supported by the IHC and ISH staining, where only few cells in the lining tissue in the treated mice notably stained for CD3 and Il17a mRNA, respectively (Figures 6 and 7). However, any observation of vast producer cells in flow cytometry was impossible due to the pre-stimulation with PMA/ionomycin. The current hypothesis is further supported by Tollenaere et al., who observed the existence of few Th17 cells in human psoriatic skin lesions which produced high levels of IL-17 cytokines (22).
The identity of major IL-17-producing cells in psoriasis has been debated in the past decade and there is evidence that neutrophils and mast cells can also be involved in IL-17 cytokine production in psoriasis, rheumatoid arthritis and infections (14, 23-25). Neutrophils were found to infiltrate the tissue lining the air pouch (Figure 5) and were also present in the air-pouch lavage fluid, but in both control and treated groups (Figure 8A). In addition, mast cells residing in the lining tissue of the air-pouch cavity have been reported (16). Hence, direct stimulation through Fcγ receptor of these innate immune cells with immune complexes might potentially result in secretion of preformed IL-17 into the air-pouch cavity (19, 24). However, there is no obvious evidence supporting granulocytes/neutrophils being the major IL-17-producing cells in the air-pouch mouse model. The proportions of GR1+IL-17A+ and GR1–IL-17A+ leukocytes (CD45+) did not reveal any particular change post treatment (Figure 8C-E) nor did anti-CD3 alone stimulated the increased secretion of IL-17 that was observed with the addition of IL-23 to the inflammatory cocktail (Figure 2). Although IL-23 receptor has been found to be expressed on neutrophils, the need for a co-stimulatory signal is a well-known feature of T-cells (26, 27).
Widely present polymorphonuclear cells in the histological sections of the tissue lining the air pouch stained negatively for Il17a mRNA in ISH (Figure 7). In addition, in agreement with Sin et al. (16), mast cells exhibited a more defined layer of cells in the lining tissue in contrast with the randomly distributed CD3+ cells which follow the same pattern as il17a-expressing cells (Figures 6 and 7). Overall, for both cell types, the current data suggest that there was no de novo synthesis of IL-17 cytokines (negative in ISH, no change in flow cytometry), nor there was vast secretion of pre-formed IL-17, which would probably have been noted as a significant reduction of the GR1+IL-17A+ population in flow cytometry. Nevertheless, more carefully designed ISH and IHC stains are required in order to evaluate the contribution of granulocytes to cytokine production in the air-pouch mouse model.
We speculate that the presence of IL-17 cytokines stimulated the production of CXCL1 by a variety of cells, such as macrophages and monocytes (28). In addition, mast cells have been shown to secrete pre-formed or newly synthesized CXCL1, and human fibroblasts can produce this chemokine upon IL-17A stimulation (29). However, there was no significant increase in the neutrophil infiltration into the air-pouch cavity throughout the time course of the studies (Figure 8A and B). This might indicate that ongoing inflammation was established in the air-pouch mouse model, resulting in already increased neutrophil infiltration, which was not drastically affected by the increase of CXCL1. Nonetheless, when other triggers, such as carrageenan, were utilized in this mouse model, it resulted in an acute inflammatory response with significant increase of polymorphonuclear cell migration into the air-pouch cavity (30).
Conclusion
The inflammatory cocktail, in a moderately viscous medium, consisting of anti-CD3, IL-23 and IL-1β, induced the secretion of IL-17A and IL-17F in the murine 6-day air-pouch model and this also triggered the downstream production of CXCL1. To our knowledge this is the first Th17-associated response established in the air pouch model.
In conclusion, the 6-day air pouch mouse model mirrors the IL-23/IL-17 axis of psoriasis-like inflammation in regard to cytokine secretion and the inflammatory reaction after the injection of anti-CD3, IL-23 with or without IL-1β, and thus this model has the potential to be used in drug discovery when targeting the Th17 inflammatory response.
Acknowledgements
The Authors highly appreciate the significant contribution of Charlotte Menne Bonefeld, who mentored us during the mouse studies. We would like to thank the important assistance of all the laboratory technicians at the Department of In vivo Biology and Safety, at LEO Pharma, as well as Kirstine Roepstorff, Lars Hornum, Paola Lovato, Josephine Hebsgaard, Britta Cathrina Martel and Jan Pravsgaard Christensen for their guidance and contribution.
Footnotes
↵Current address: Paul-Ehrlich-Institute, Research Group of Molecular Biotechnology and Gene Therapy, Langen, Germany
This article is freely accessible online.
Authors’ Contributions
Conceptualization: JK; methodology: JK, DSMD and FTC; formal analysis: FTC; investigation: FTC and DSMD; writing – original draft: FTC and DSMD; visualization: FTC; writing – review and editing: JK; funding acquisition: JK; resources: JK; supervision: JK.
Conflicts of Interest
The Authors declare that no competing of interests exist.
- Received March 13, 2021.
- Revision received April 16, 2021.
- Accepted April 22, 2021.
- Copyright © 2021 International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.