


Abstract
Background: Amrubicin hydrochloride is administered as second- or third-line therapy for small cell lung cancer, and is known to cause severe myelotoxicity. This study evaluated the efficacy and safety of weekly amrubicin for refractory/relapsed small cell lung cancer. Patients and Methods: A single-arm, open-label, multicenter, phase II study of weekly amrubicin was performed in 21 patients at seven centers in Japan from 2012 through 2015. Results: A partial response (PR) was noted in one out of the first 18 patients. The study was terminated early according to the termination criteria in the protocol. In total, the response rate was 19% (no complete responses and four PRs) and the disease control rate was 81% (17/21). Median overall survival was 288 days (95% confidence interval(CI)=208-424 days), while median progression-free survival was 113 days (95% CI=45-202 days). Conclusion: This study failed to demonstrate any efficacy of weekly amrubicin for refractory/relapsed small cell lung cancer.
Small cell lung cancer accounts for about 15% of all lung cancers and is known to have a very poor prognosis. Despite advances in chemotherapy, patients with advanced small cell lung cancer still have a median survival time of 9 to 12 months and a 2-year survival rate of about 5-20%. Accordingly, development of more effective chemotherapy is required.
Amrubicin hydrochloride was developed in Japan and is a derivative of the anthracycline doxorubicin hydrochloride. In Japan, Europe, and the United States, it has been reported that amrubicin hydrochloride has a good antitumor activity against small cell lung cancer (1-5). It is frequently administered alone as second-line or third-line therapy for small cell lung cancer, but is known to cause severe myelotoxicity.
In clinical practice, amrubicin hydrochloride is usually administered once daily for 3 consecutive days, every 3 weeks (1). For other medications, it has been suggested that toxicity can be reduced to a low level without attenuating efficacy by dividing the dose and performing weekly administration (6-8). A weekly dosing regimen of amrubicin hydrochloride might reduce side-effects including myelosuppression, which is severe with the conventional regimen. As a result, there is a possibility that the dose intensity and effectiveness of treatment could be improved. It is also possible that the weekly regimen may be more convenient for patients by reducing the number of hospital visits. Therefore, as a new administration regimen for amrubicin hydrochloride in patients with previously treated refractory or relapsed lung cancer, weekly administration was assessed in a dose-finding phase I study. As a result, it was concluded that the maximum tolerated dose was 65 mg/m2 and the recommended dose was 60 mg/m2 (9). In order to further investigate the efficacy and safety of weekly amrubicin hydrochloride as second-line therapy for refractory or relapsed small cell lung cancer, we planned a phase II study using the recommended dose from the phase I study.
Eligibility criteria for study patients.
In this phase II study, the efficacy and safety of weekly amrubicin therapy was assessed for small cell lung cancer that was refractory to first-line chemotherapy or had relapsed.
Patients and Methods
Patients. Patients who satisfied the eligibility criteria (Table I) were enrolled. Refractory relapse was defined as no response to first-line chemotherapy or relapse within 90 days after the date of the final dose of first-line chemotherapy, while sensitive relapse was defined as relapse 90 days or more after the date of the final dose of first-line chemotherapy.

Participant flow chart.
Trial design and treatment. This was a single-arm, open-label, multicenter, phase II study. It was registered with the UMIN Clinical Trials Registry (UMIN-CTR, URL: http://www.umin.ac.jp/ctr/): registration number UMIN000009440. The study protocol was reviewed and approved by each Institutional Review Board, and written informed consent was obtained from all of the patients. Amrubicin was administered at a dose of 60 mg/m2 on days 1 and 8, and this regimen was repeated every 3 weeks until disease progression or development of intolerable toxicity.
Endpoints. The primary endpoint was the response rate as defined by the Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1 (10). Confirmed partial response (PR) was defined as PR also on the second computed tomographic evaluation at 4 weeks or more after the initial detection of PR. Secondary endpoints were progression-free (PFS) and overall (OS) survival, and the incidence of adverse events. PFS was defined as the time from the day of enrollment to the day that disease progression was confirmed by imaging studies or the day of death from any cause, whichever came first. OS was defined as the time from the day of enrollment to the day of death from any cause, and patients were censored on the last date of survival was confirmed. Adverse events were monitored and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 4.0 (11).
Demographic characteristics of study participants.
Statistical analysis. By assuming that efficacy would be equal to or higher than that achieved with conventional administration (2, 3), and assuming that the threshold response rate was 20%, the expected response rate was 40%, α=0.05 (one-sided), and β=0.2, it was calculated that 18 patients were required for the first stage of the Simon two-stage theory (Minimax model). If there were at least five responders among the initial 18 patients, 15 more patients would be added at the second stage. If 11 responders were obtained among the total of 33 patients, this regimen would be judged to be effective. In consideration of possible inappropriate enrollments, two more patients were added, and the target number of patients was therefore set as 35.
According to the protocol, this therapy would be defined as ineffective if a response of PR or better was achieved by fewer than five of the initial 18 patients, resulting in termination of the study. The response rate, PFS, OS, and safety (incidence of adverse events) were analyzed.
Results
Twenty-one patients from seven centers in Japan were enrolled during the period from December, 2012 through May, 2015. The participant flow chart is shown in Figure 1.
Baseline data are listed in Table II. Twenty-one patients were registered, among whom 18 were men. Their median age was 67 years (range=49-81 years). All patients had a history of smoking. At initial diagnosis, only five patients had limited disease. The tumor showed refractory recurrence in four patients and sensitive recurrence in 17.
Efficacy of amrubicin hydrochloride in study patients.
PR was achieved in one out of first 18 patients. As recommended by the independent Data and Safety Monitoring Committee, the trial was terminated because it did not satisfy the criteria (PR in five or more of the first 18 patients) for proceeding to the second step according to the Simon two-stage theory.
The number of cycles administered ranged from 1 to 8 and the median number was 2. The response rate was 19% (Table III). There was no patient with a complete response (CR) and four patients with PR. The disease control rate (PR + CR + SD/intention to treat population) was 81% (17/21). The median survival time was 288 days (Figure 2, 95% confidence interval=208-424 days), and median PFS was 113 days (Figure 3, 95% CI=45-202 days).
A grade 3-4 decrease of the white blood cell count and absolute neutrophil count occurred in 11 (55%) and 15 patients (75%), respectively, while febrile neutropenia was found in four patients (20%). Grade 3-4 anemia or thrombocytopenia was noted in eight patients (40%) and three patients (15%), respectively. Grade 3-4 elevation of aspartate transaminase, alanine transaminase, or creatinine occurred in one patient each (5%), and grade 3-4 hyponatremia was observed in 3 patients (15%). As non-hematological toxicities, grade 3-4 dyspnea and oral mucositis were found in one (5%) and two patients (10%), respectively. In addition, there were two cases (10%) of grade 2 pneumonitis and 1 case (5%) of grade 3 pneumonitis. There were no treatment-related deaths (Table IV).
Discussion
This study was terminated early due to the low response rate and further assessment of weekly administration of amrubicin was stopped. Inoue et al. reported a response rate of 67% for amrubicin in patients with sensitive relapse (12). Although 81% of the patients in our trial had sensitive relapse, the response rate was only 19% and we were unable to obtain a response rate as high as that of Inoue et al. Although the response rate of 19% was below expectations, weekly amrubicin achieved a high disease control rate, since 81% of the patients had stable disease or PR. Therefore, our results indicate that weekly administration of amrubicin hydrochloride was able to stabilize the disease, but was not as effective as the conventional regimen for obtaining responses.
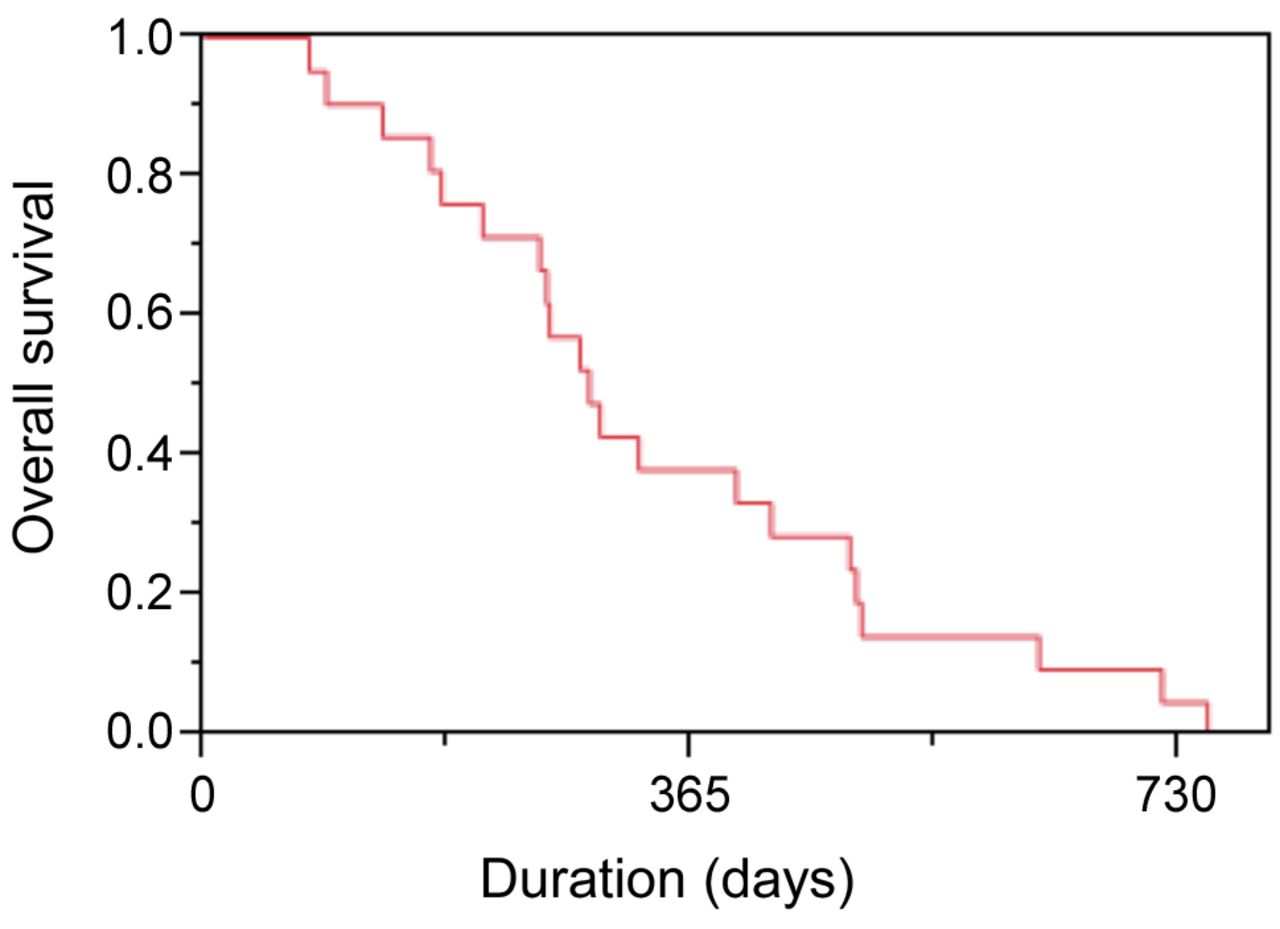
Overall survival of study patients on amrubicin hydrochloride.

Progression-free survival of study patients on amrubicin hydrochloride.
Discontinuation of this trial might have been avoided if the disease control rate had been selected as the primary endpoint instead of the response rate. A possible reason for the low response rate in this study is that etoposide, which is a topoisomerase II inhibitor like amrubicin hydrochloride and which can induce cross-resistance, might have been used in prior treatment for many patients, as reported previously (13).
Adverse events experienced by study patients on amrubicin hydrochloride.
Our intention was to reduce the frequency of adverse events, especially febrile neutropenia, which sometimes leads to treatment-related death, by modifying the treatment schedule to weekly administration from conventional administration on three consecutive days. However, 20% of the patients in this study developed febrile neutropenia and we were unable to show that weekly administration was safer than the conventional regimen. In conclusion, this study failed to demonstrate that weekly amrubicin has the same efficacy as conventional amrubicin therapy and no further development of this new regimen is warranted.
Acknowledgements
The Authors thank all of the patients and their families, as well as the investigators who participated in this study.
Footnotes
This article is freely accessible online.
- Received August 2, 2018.
- Revision received September 28, 2018.
- Accepted October 1, 2018.
- Copyright© 2018, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.