


Abstract
Background/Aim: Novel information on the role of endogenous compounds in regulating physiological and pathological process are of interest, as it may lead to the development of better strategies for disease management. The role of angiotensin II and the signaling of type 1 angiotensin II receptor (AGT1R) in T-lymphocyte activation and interleukin-2 (IL-2) production are largely unknown. Materials and Methods: Jurkat T-cells were treated with AGT1R inhibitor candesartan and stimulated with phorbol myristate acetate (PMA) and ionomycin. T-Cell activation, associated cytokine production and levels of signaling proteins were evaluated by flow cytometry and western blot analysis. Results: Candesartan significantly suppressed PMA and ionomycin-induced CD25 expression and IL-2 production. Regarding the molecular mechanism involved, we showed that such suppressive effects of blocking of AGT1R by candesartan resulted in the significant inhibition of ERK activation in PMA-stimulated Jurkat T-cells. The effect of ERK inhibition on T-cell activation was further confirmed. Treatment with FR180204, a specific ERK inhibitor, reduced T-cell activation and IL-2 secretion. Conclusion: AGT1R signaling is essential for T-cell activation and IL-2 production, and the inhibition of this pathway suppressed T-cell activation via an ERK-dependent mechanism.
The most fundamental trait of endogenous mediators is their activity in regulation of cell, tissue and organ functions. The dysregulation of, or altered response to such mediators may lead to the pathogenesis of diseases. The novel actions of known endogenous substances, including hormones, have been intensively unraveled and such information is of importance for the better understanding of biology, and physiological and pathological processes. Regarding the cardiovascular system, the renin–angiotensin system (RAS) is essential for regulation of blood pressure and fluid homeostasis (1, 2). The primary effector molecule, angiotensin II, acts through type 1 and type 2 angiotensin II receptors (AGT1R and AGT2R).
There is evidence indicating that angiotensin II may play a role in regulating the immune system (3-6). For example, angiotensin II was shown to stimulate production and secretion of several pro-inflammatory cytokines involved in the pathogenesis of atherosclerosis (7) and chronic kidney injury (5). In addition, study in an in vivo model of immune-mediated renal injury revealed that in mice the deletion of Agt1r reduced glomerular expression of monocyte chemoattractant protein-1 and ameliorated proteinuria as well as glomerular tissue damage (8). More importantly, a classic angiotensin receptor blocker has been proposed to reduce inflammation and modulate autoimmunity (9, 10).
As knowledge on the direct regulatory effect of angiotensin II in controlling immune cell functions and AGT1R signaling in immune cells remains limited, the present study aimed to provide more information regarding the role of angiotensin II and the function of AGT1R in the activation of T-lymphocytes (T-cells).
Materials and Methods
Materials and reagents. Jurkat T-cells, clone E6-1, were obtained from the American Type Culture Collection (Manassas, VA, USA). Roswell Park Memorial Institute (RPMI)-1640, fetal bovine serum (FBS), L-glutamine, and penicillin and streptomycin were purchased from Gibco/Life Technologies (Carlsbad, CA, USA). Selective AGT1R inhibitor candesartan, pretein kinase C activator phorbol 12-myristate 13-acetate (PMA), and calcium ionophore ionomycin calcium salt were purchased from Sigma Chemical (St. Louis, MO, USA). Cell proliferation reagent WST-1, and AKT inhibitor perifosine were also from Sigma Chemical. Lysis buffer and Complete Mini cocktail protease inhibitor were obtained from Roche Molecular Biochemicals (Indianapolis, IN, USA). BCA protein assay kit and SuperSignal West Pico Chemiluminescent substrate were from Pierce Biotechnology (Rockford, IL, USA). Primary antibodies for extracellular signal-regulated kinase (ERK), p-ERK, AKT, and p-AKT and peroxidase-conjugated secondary antibodies were from Cell Signaling Technology (Danvers, MA, USA). Fluorescein isothiocyanate (FITC)-conjugated monoclonal antibody for CD25 (CD25-4E3) was purchased from Thermo Scientific (Fremont, CA, USA). ERK inhibitor, FR180204, was from Merck Millipore (Darmstadt, Germany) and an enzyme-linked immunosorbent assay (ELISA) system was obtained from R&D Systems (Minneapolis, MN, USA).
Cell culture. Jurkat T-cells were cultivated in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% FBS, 2 mM L-glutamine, and 100 U/ml penicillin and streptomycin. Cells were cultured in 24-well plates and maintained in a 37°C humidified incubator with 5% CO2.
Cytotoxicity assays. To evaluate cytotoxicity of candesartan, cells were seeded onto 96-well plates at a density of 5×105 cells/well and culture medium with different concentrations of candesartan was added to obtain final concentrations of 0-10 μM. After 24 h, cell viability was measured using WST-1 reagent according to the manufacturer's protocol. The percentage of cell viability was calculated as the absorbance of treated cells relative to that of untreated cells.
Nuclear staining assay. Cells were seeded at a density of 1×105 cells per well onto a 96-well plate and treated with 0-1 μM candesartan for 24 h. Apoptotic and necrotic cell death was analyzed by subsequent incubation with 10 μg/ml of Hoechst 33342 and 5 μg/ml of propidium iodide (PI) for 30 min at 37°C. Nuclear condensation and DNA fragmentation of apoptotic cells, as well as PI-positive necrotic cells, were visualized under a fluorescence microscope to observe nuclear morphology and PI-positive necrotic cells (Olympus IX51 with DP70 Digital camera system; Olympus, Central Valley, PA, USA).
Western blot analysis. Jurkat T-cells (2×106) were cultured in a 24-well plate. Cells were pre-treated with 0 or 1 μM of candesartan for 15 min then stimulated with 25 ng/ml PMA plus 1 μM ionomycin for 4 and 24 h. After treatment, cells were harvested and solubilized for 1.5 h at 4°C in lysis buffer containing protease inhibitor mixture. Protein content in cell lysates was quantified using BCA protein assay kit. Equal amounts of denatured protein samples (40 μg) were loaded onto 10% sodium dodecyl sulfate–polyacrylamide gel to undergo electrophoresis for separation of proteins by their molecular weight. The separated proteins were then transferred onto 0.45-μm nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). The membranes were blocked with 5% non-fat dry milk in 25 mMTris•HCl (pH 7.5), 125 mM NaCl, and 0.05% Tween 20 (TBST) for 2 h prior to incubation overnight at 4°C with the primary antibodies specified above. The membranes were washed three times with TBST and incubated with appropriate horseradish peroxidase-labeled secondary antibodies for 2 h at room temperature. The immune complexes were detected by chemiluminescent substrate and quantified using ImageJ 1.51j8 software (National Institutes of Health, Bethesda, MD, USA).
Flow cytometric analysis. Cells were pre-treated with either 1 μM of candesartan, 10 μM of FR180204, or 10 μM of perifosine for 15 min, followed by 25 ng/ml PMA plus 1 μM ionomycin. Cells stimulated with PMA and ionomycin without pre-treatment served as control. After 24 h, cells were washed with phosphate-buffered saline twice and incubated with FITC-conjugated monoclonal anti-CD25 for 30 min at 4°C. After staining, cells were washed twice with phosphate-buffered saline and the percentage of cells expressing CD25 was quantitated using a Guava easyCyte flow cytometer (Merck Millipore, Darmstadt, Germany).
Cytokine quantification. Cells were cultured at density of 2×106 cells/ml in a 24-well plate. The cells were pre-treated with either 1 μM of candesartan, 10 μM of FR180204, or 10 μM of perifosine for 15 min and stimulated with 25 ng/ml PMA plus 1 μM ionomycin for 5 h. Culture supernatants were collected and analyzed for IL-2 concentration using an ELISA assay, according to the manufacturer's protocols.
Statistical analysis. The data are presented as the mean±standard error (SEM) from three to five independent experiments. Statistical analysis was determined using two-way ANOVA and a post hoc test at a significance level of p<0.05.
Results
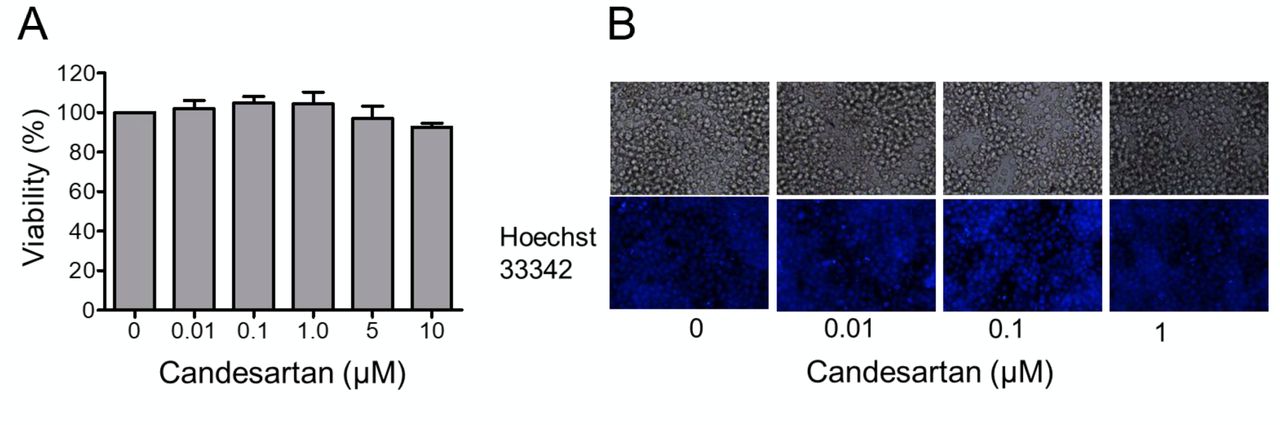
Effects of AGT1R blocker, candesartan on the viability of Jurkat T-cells. To study the role of AGT1R in regulation of T-lymphocyte activation, a selective AGRT1R inhibitor, candesartan, was used. Firstly, the cytotoxic effect of candesartan on the cells was determined. Jurkat T-cells were treated with serial dilution of candesartan ranging from 0 to 10 μM for 24 h. Cell viability was then measured using a WST-1 assay. Figure 1A shows that there was no significant difference in cell viability observed in response to the treatment of candesartan at 0-10 μM. This was confirmed by Hoechst 33342/PI staining assay showing that there were no apoptotic cells observed in cells treated with 1 μM candesartan (Figure 1B).
Blocking of AGT1R by candesartan suppressed PMA/ionomycin-induced Jurkat T-cell activation. A previous study has shown that T-lymphocytes express AGT1R and produce local angiotensin II, which modulates T-cell functions (11) Therefore, we sought to determine the effects of AGT1R blockade, using candesartan, on the activation of T-cells. Jurkat T-cells were treated with the known T-cell activator, PMA (25 ng/ml) and ionomycin (1 μM) in the presence or absence of candesartan (1 μM) for 24 h. The expression of T-cell activation marker CD25 was then analyzed by flow cytometry. As shown in Figure 2A, unstimulated Jurkat T-cells expressed a low level of CD25. The addition of candesartan significantly reduced CD25 expression by 32% in stimulated cells (16.7%±0.3 in PMA/ionomycin vs. 11.2%±0.5 in PMA/ionomycin plus candesartan) (Figure 2B).
As we observed a significant decrease in T-cell activation marker in response to candesartan treatment, we sought to determine whether blocking AGT1R in Jurkat T-cells also inhibited their cytokine production. Jurkat T-cells were pre-treated with candesartan and stimulated with PMA and ionomycin for 5 h. Culture supernatant was then collected and analyzed for IL-2 concentration using ELISA. Pharmacologically blocking AGT1R was found to significantly suppress IL-2 production in activated T-cells (Figure 2C).
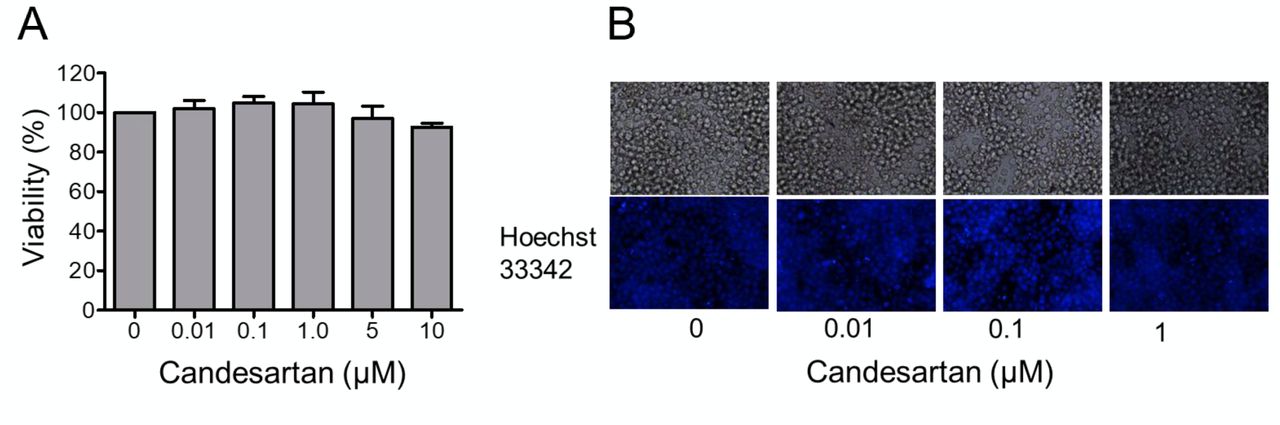
Effects of candesartan on the viability of Jurkat T-cells. A: Jurkat T-cells were treated with different concentrations of candesartan (0-10 μM) for 24 h. Cell viability was then determined using a WST-1 assay. Data are the mean±SEM (n=3). B: Hoechst33342/propidium iodide (PI) co-staining for nuclear morphology of the cells after treatment for the indicated time.
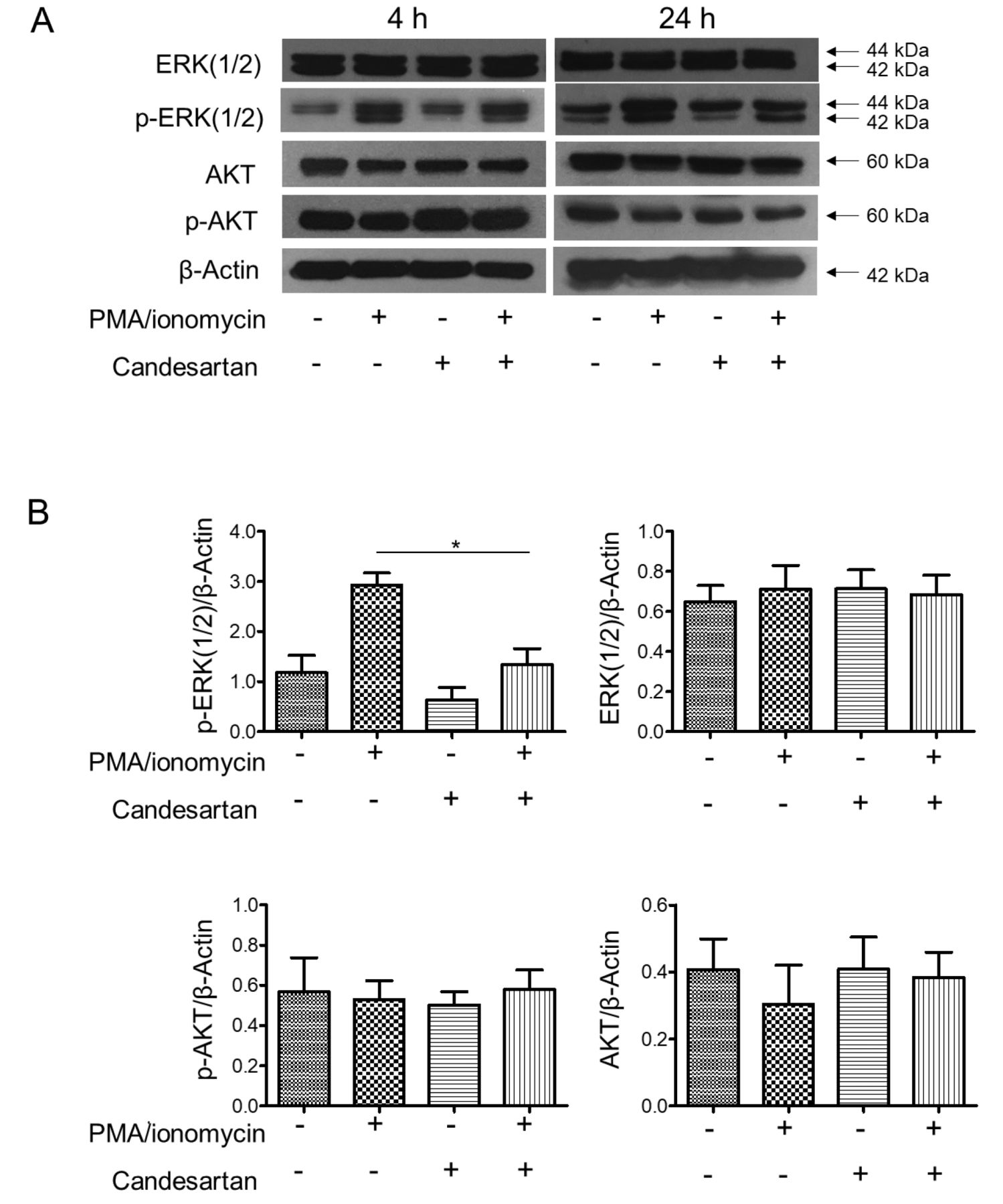
Candesartan inhibits T-cell activation through ERK suppression. Having shown that treatment with candesartan inhibited Jurkat T-cell activation and IL-2 production, we therefore investigated the possible mechanism by which inhibition of AGT1R could affect T-cell activation. Expression of ERK and AKT proteins, actors in known cellular pathways regulating T-cell activation, was then determined by western blot analysis in PMA/ionomycin-activated cells treated with candesartan. As shown in Figure 3A, stimulation of the cells with PMA and ionomycin markedly induced ERK phosphorylation in Jurkat T-cells; however, pre-treatment with candesartan significantly attenuated this activation (p-ERK(1/2)) (Figure 3B). We observed that AKT activation was not altered in this experiment, suggesting that the AKT pathway was not affected by AGT1R blockade.
In order to confirm the role of ERK activation in T-cell activation and IL-2 secretion, Jurkat T-cells were pre-treated with the known ERK inhibitor FR18024 prior to PMA/ionomycin stimulation. The inhibition of ERK phosphorylation in this way significantly suppressed CD25 expression (Figure 4A) and IL-2 production (Figure 4B) in Jurkat T-cells, confirming our finding above. Moreover, treatment of the cells with the AKT inhibitor perifosine, did not alter T-cell activation induced by PMA and ionomycin stimulation. These results confirmed that the blocking of AGT1R attenuated T-cell activation via suppression of an ERK-related mechanism.
Discussion
Although since long known for its role in regulating hemodynamic homeostasis and water balance (2), novel biological and physiological functions of angiotensin II have been continuously discovered. For instance, angiotensin II was recently shown to have a function in enhancing cancer stem cell-like phenotypes in lung cancer cells (12). Exposure of lung cancer cells to angiotensin II at non-toxic concentrations induced key features of cancer stem cells, including their ability to grow as 3D tumor spheroids and under anchorage-independent conditions, together with the dramatic increase of emission of cancer stem cell markers (12). Angiotensin II was also found to affect cell proliferation (13-15), as well as the production and secretion pattern of pro-inflammatory cytokines (16-18). Such information leads to the possibility that angiotensin II may play a role in controlling cell signaling in inflammation and immune-mediated diseases.
T-Lymphocytes express AGT1R and other RAS components and locally produced angiotensin II has been shown to regulate immune cells (19, 20). Angiotensin II infusion of immunodeficient mice lacking T-and B-lymphocytes reduced hypertension and vascular injury, while adoptive transfer of T-cells reversed these effects (21), suggesting the role of AGT1R signaling in the development of vascular diseases independent of hemodynamic function. Furthermore, in autoimmune disease models, including multiple sclerosis, and rheumatoid arthritis, infusion of angiotensin II led to severe disease while treatment with RAS inhibitor ameliorated the severity of the diseases (10, 22, 23), suggesting crucial roles of angiotensin II as an proinflammatory mediator and immune regulator. However, how angiotensin II and AGT1R are involved in regulating T-cell function is still not clear.
Here we reported the role of AGT1R in the activation of Jurkat T-cells using a specific AGT1R blocker, candesartan. Our current data reveal that candesartan significantly suppressed the activation of T-cells as shown by CD25 expression and IL-2 production in stimulated T-cells (Figure 2B and C). For a mechanistic explanation of these effects, we found that blocking AGT1R resulted in significant depletion of ERK phosphorylation and consequently limited the activation of the cells. Our finding is consistent with the fact that not only is the ERK-related mechanism critical for T-cell activation (24), but also that this pathway is important for the growth and survival of CD8+ T-cells (25). Regarding IL-2 production, it was shown that ERK regulates the production and secretion of IL-2, as the transfection of Jurkat T-cells with a dominant negative mutant of ERK1 resulted in diminished IL-2 production in stimulated cells (26).
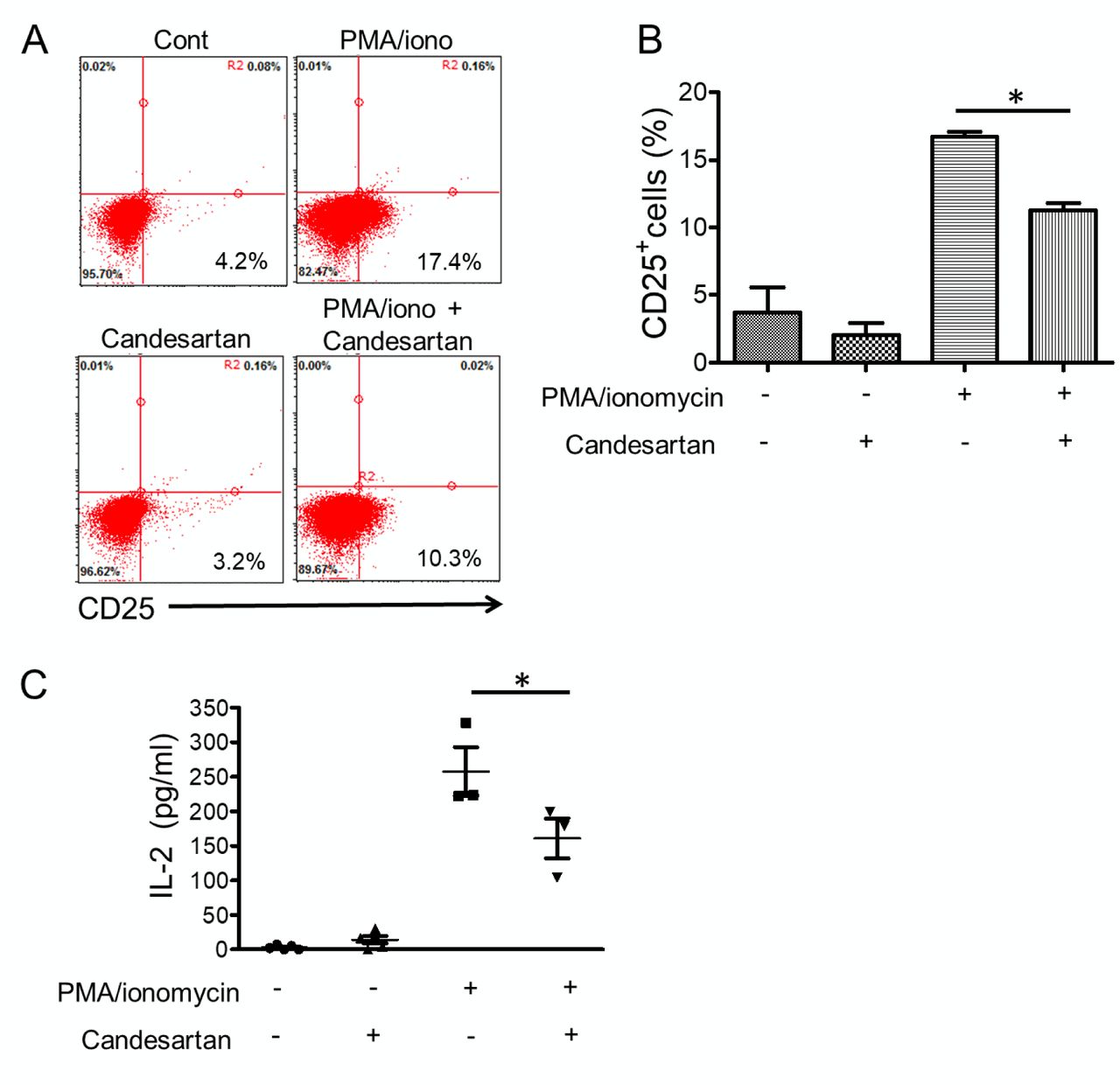
Type 1 angiotensin II receptor inhibitor candesartan reduced T-cell activation. A: Representative dot plot of Jurkat T-cells stained for CD25 expression. Jurkat T-cells were pre-treated with candesartan 1 μM and stimulated with 25 ng/ml of a protein kinase C activator, phorbol myristate acetate (PMA), plus 1 μM of a calcium ionophore, ionomycin. After 24 h, cells were harvested and stained with fluorescein isothiocyanate-conjugated antibody to CD25 for flow cytometric analysis, as described in the Materials and Methods. B: The bar graph shows the percentage of CD25+-expressing cells. C: Jurkat T-cells were treated with 1 μM candesartan and stimulated with 25 ng/ml PMA and 1 μM ionomycin for 5 h. Culture supernatant was collected and analyzed for interleukin-2 (IL-2) level using an enzyme-linked immunosorbent assay. Data are the mean±SEM (n=3). *Significantly different at p<0.05.
Excessive T-cell activation and IL-2 production are known as key players in several pathological processes. IL-2, predominantly produced by activated T-cells, acts as T-cell growth factor to promote cell proliferation and secretion of pro-inflammatory cytokines such as Interferon-γ, and tumor necrosis factor-α (27). In systemic lupus erythematosus, serum levels of IL-2 and soluble IL-2 receptor are increased in patients with active disease (28, 29). In addition, enhanced immune activation by depletion of suppressive regulatory T-cells (Tregs) aggravates aortic aneurysm induced by angiotensin II (30). Thus, adoptive transfer of Tregs to the Treg-deficient mice was found to rescue the animals from increased susceptibility to aortic rupture (30). This is consistent with a study showing that immunosuppression by in vivo expansion of Tregs reduced angiotensin II-induced aortic stiffening (31).
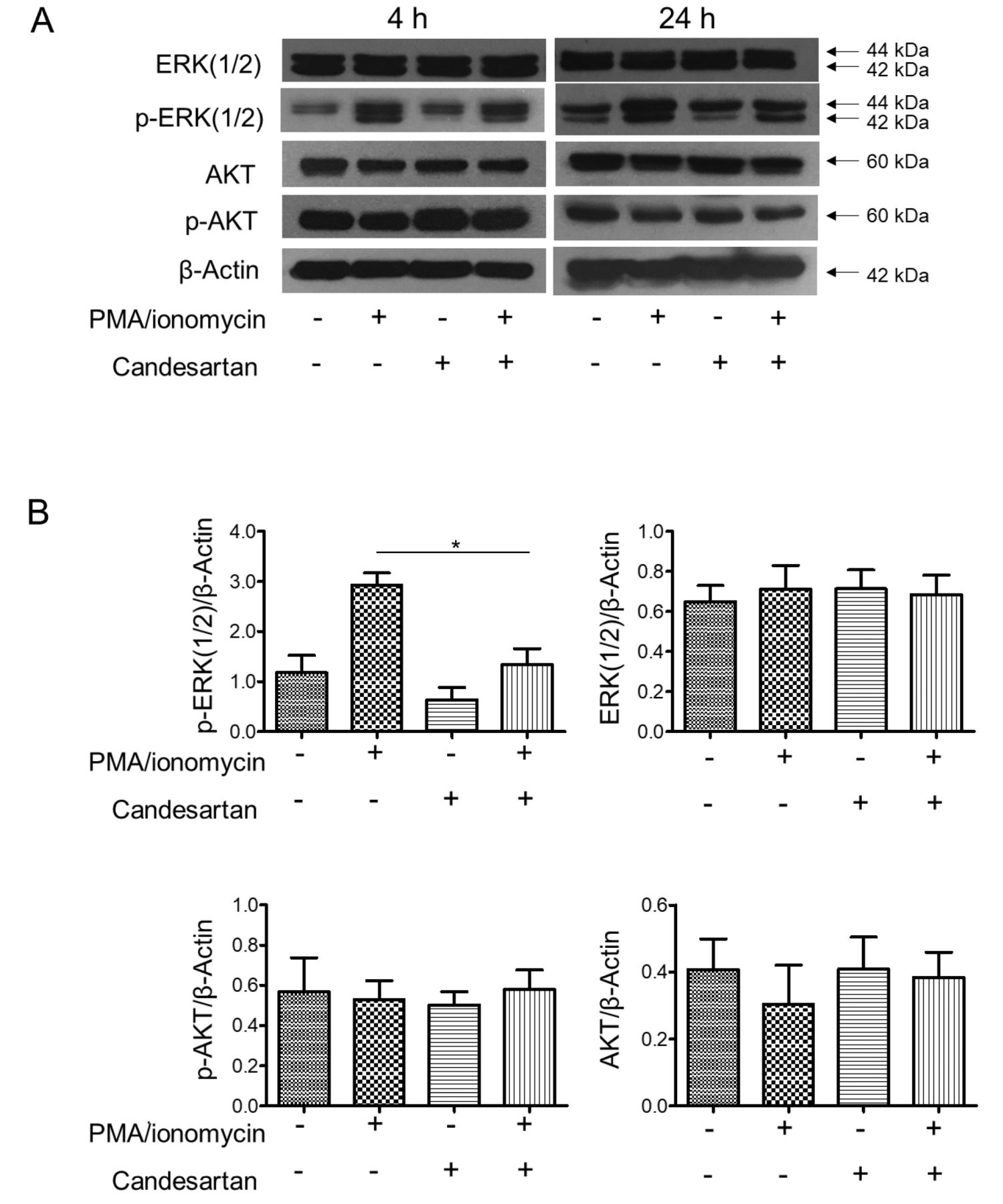
Candesartan inhibited the activation of extracellular signal-regulated kinase (ERK) in Jurkat T-cells. A: Jurkat T-cells were pre-treated with 1 μM candesartan and stimulated with 25 ng/ml of an protein kinase C activator, phorbol myristate acetate (PMA) plus 1 μM of a calcium ionophore, ionomycin for 4 and 24 h. Cells were lysed and soluble lysates were analyzed by western blotting for the expression of total ERK(1/2), activated ERK (pERK1/2), total AKT, and activated AKT (pAKT). β-Actin was used to confirm equal protein loading. Data shown are representative of three independent experiments. B: Quantification of target-protein expression normalized with β-actin relative to the expression of non-treated control. Data are the mean±SEM (n=3). *Significantly different at p<0.05.
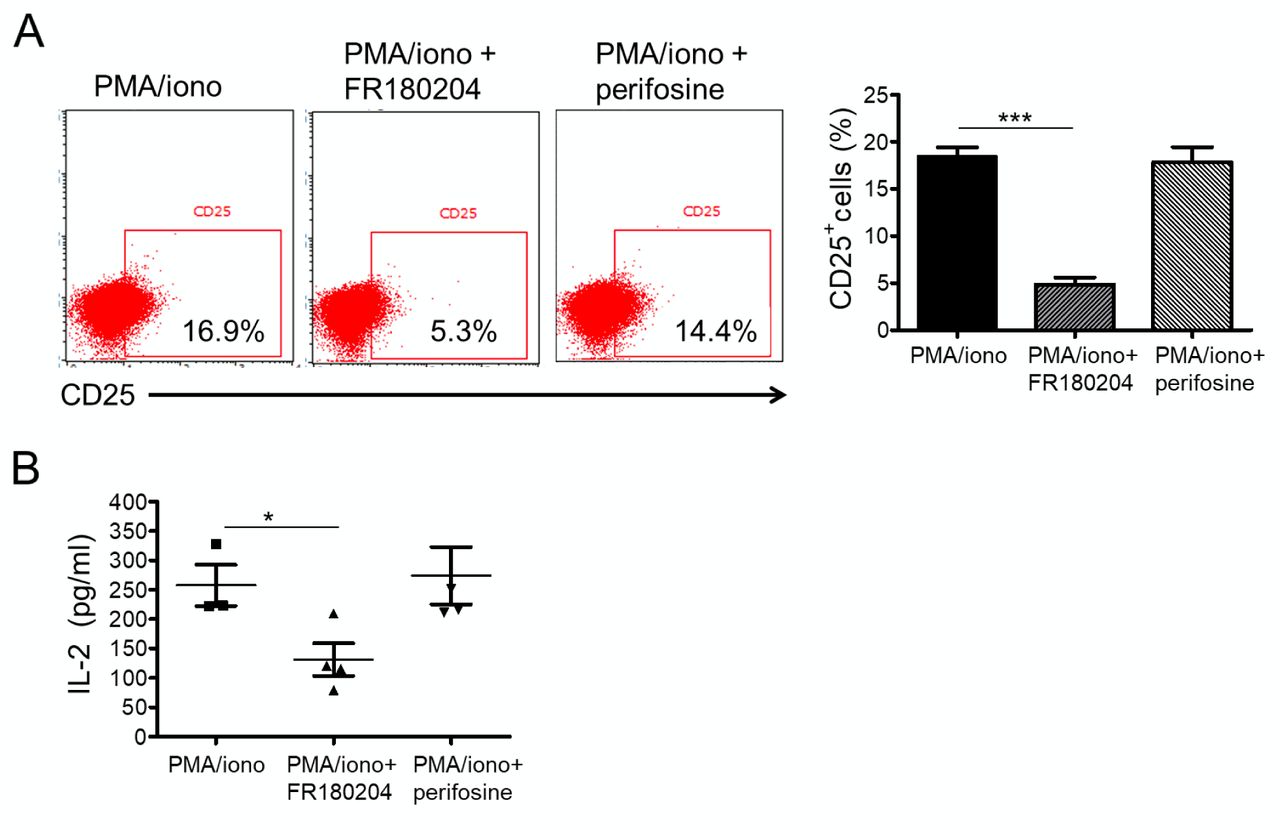
T-Cell activation is dependent on the activation of extracellular signal-regulated kinase (ERK), but not AKT pathway. A: Dot plots represent CD25 expression of Jurkat T-cells. Jurkat T-cells were pre-treated with either 10 μM of an ERK inhibitor, FR180204 10 μM of an AKT inhibitor, perifosine for 15 min. The cells were then stimulated with 25 ng/ml phorbol myristate acetate (PMA) plus 1 μM ionomycin. After 24 h, cells were harvested and stained with a fluorescein isothiocyanate-conjugated antibody to CD25 for flow cytometric analysis. The bar graph shows the percentage of cells expressing CD25. Data are the mean±SEM (n=3). Significantly different at *p<0.05, and ***p<0.001. B: Jurkat T-cells were treated with either 10 μM FR180204 or 10 μM perifosine and stimulated with 25 ng/ml PMA plus 1 μM ionomycin for 5 h. Culture supernatant was collected and analyzed for interleukin-2 (IL-2) levels using an enzyme-linked immunosorbent assay. Data are the mean±SEM (n=3). *Significantly different at p<0.05.
Our study revealed that signaling through AGT1R plays a role in regulation of T-lymphocyte activation and IL-2 secretion. The blockage of AGT1R resulted in the decrease of ERK phosphorylation, highlighting the significant role of ERK in control of T-cell activation and IL-2 production. This knowledge may benefit the better understanding of the role of angiotensin II and its signaling on immune cell function.
Acknowledgements
This research was funded by Chulalongkorn University, CU-GR_ 60_45_33_10.
Footnotes
This article is freely accessible online.
- Received June 19, 2018.
- Revision received June 29, 2018.
- Accepted July 11, 2018.
- Copyright© 2018, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.