


Abstract
Prostate cancer antigen (PCA)-1/AlkB homologue 3 (ALKBH3) has been identified as a clinically significant factor and siRNA of PCA-1 inhibits DU145 proliferation both in vitro and in vivo. HUHS015 (1), a previous reported PCA-1 small-molecule inhibitor, was also effective without any obvious side-effects or toxicity. The potency of HUHS015, however, is not satisfying. We thought the reason is poor solubility of HUHS015 because insoluble material remained at the injection site after subcutaneous administration. To improve this inhibitor's solubility, we prepared various salts of HUHS015 and examined their solubility, which resulted in the selection of HUHS015 sodium salt (2) for further studies in vivo. Next, we compared the pharmacokinetics of 1 and 2 via several administration routes. We observed significant improvements in the pharmacokinetic parameters. For example, subcutaneous administration of 2 increased the area under the curve (AUC)0-24 by 8-fold compared to 1 and increased the suppressive effect on the proliferation of DU145 cells in a xenograft model.
Prostate cancer is extremely serious when malignant and developing novel anti-prostate cancer drugs that are effective against both androgen-dependent and independent types is necessary (1). Tsujikawa et al. reported that a novel gene encoding a DNA and/or RNA-alkylating damage-repair enzyme, named prostate cancer antigen (PCA)-1 (2) or AlkB homologue 3 (ALKBH3), that is often highly expressed in clinical prostate cancer cells. Patients with high levels of PCA-1 expression tend to shift into androgen-independent types, and PCA-1 is an important factor for prognosis (2). Furthermore, genetic inhibition of PCA-1 with siRNA effectively inhibits the growth of androgen-independent prostate cancer cells, such as DU145, which express high levels of PCA-1 (3-4). In addition, loss of PCA-1 leads to 3-methylcytosine accumulation and reduces cell proliferation in various cell lines (5). Therefore, a small, orally-available PCA-1 inhibitor would be a novel and clinically effective anti-prostate cancer drug, even for hormone-independent cancers. We designed and synthesised a novel small PCA-1 inhibitor, 1-(5-methyl-1H-benzimidazol-2-yl)-4-benzyl-3-methyl-1H-pyrazol-5-ol, HUHS015 (1) (Figure 1), based on a random screening assay using recombinant PCA-1 (6). HUHS015 demonstrated potent inhibition of the enzymatic activity of PCA-1 (half maximal inhibitory concentration (IC50)=0.67 μM) and significantly suppressed the growth of DU145 cells both in vitro (IC50=2.2 μM, 0.7 μg/ml) and in a mouse xenograft model at 32 mg/kg (subcutaneous administration) (6). However, in vivo, the inhibitory potency of HUHS015 was not satisfactory. We hypothesised that the weaker potency of HUHS015 in vivo is due to its poor solubility, because insoluble material remains, including HUHS015, at the injection area. Therefore, in this study we prepared several salts of HUHS015 and identified the sodium salt (2) as a suitable compound for in vivo studies. We demonstrated that the increased solubility of 1 improves its bioavailability and suppression of DU145 tumor growth in a mouse xenograft model.
Materials and Methods
Synthesis of 1-(5-methyl-1H-benzimidazol-2-yl)-4-benzyl-3-methyl-1H-pyrazol-5-ol (HUHS015). The synthesis of HUHS015 (Figure 1) was described in a previous study (6).
Cell culture. The human prostate cancer cell line DU145 was supplied from Riken BioResource Center (Tsukuba, Japan). The cell line was maintained in RPMI1640 supplemented with 10% foetal bovine serum, 100 units/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified atmosphere containing 5% CO2.
Animal care. Male nude BALB/c mice and male Sprague-Dawley (SD) rats were purchased from Japan SLC Inc. (Shizuoka, Japan). The animals were housed under conditions of constant temperature and humidity and fed a standard diet and water ad libitum. Our Institutional animal care committee approved all animal experiments.
Sample preparation of 1 and 2. Compounds 1 and 2 were prepared at 32 mg/5 ml for oral administration (p.o.) and 32 mg/1 ml for subcutaneous (s.c.) and intraperitoneal (i.p.) administration. Compound 2 was also prepared at 0.25 and 0.5 mg/1 ml for intravenous (i.v.) administration. For p.o. and s.c., compound 1 was suspended in 0.5% Methyl cellulose (0.5% MC). For p.o., s.c. and i.p. administration, 2 was suspended in 0.5% MC or dissolved with Polyethylene glycol300 (PEG300) and an equal mol of 1 N NaOH. Compound 2 was also dissolved in a 50% volume of Dimethyl sulfoxide (DMSO) and an equal mol of 1 N NaOH. Next, 15% volume of EtOH and H2O was added. For i.v. administration, 2 was dissolved in a 50% volume of PEG300 and an equal mol of 1 N NaOH, followed by the addition of H2O.
Measurement of serum concentration of HUHS015 in rats. Male SD rats were fed ad libitum for the i.v., s.c. and i.p. tests. The rats were fasted for 18 h before the p.o. test. After the administration of each route, 5, 15, 30 min, 1, 2, 6 or 24 h later, blood samples were collected with heparin, as an anti-coagulant, from the abdominal vein under anaesthesia. The serum was then separated by centrifugation and all samples were stored at −35°C until analysis. The concentration of each sample was determined using high-performance liquid chromatography (HPLC).
Growth inhibition of DU145 xenograft in mice. Male nude BALB/c mice were implanted subcutaneously with approximately 100 μl 8×106 DU145 cells mixed with BD Matrigel™ Basement Membrane Matrix High Concentration (BD Biosciences #354248, Two Oak Park, Bedford, MA 01730) into the right flank. When the estimated tumour volume reached 255 to 573 mm3, (100-300 mm in our previous study(6)), the animals were divided into 3 experimental groups of 3-4 mice and treated subcutaneously with 32 mg/kg of 1 or 2 suspended in 0.5% MC for 13 days once per day. The tumor volume was calculated on days 0, 2, 3, 4, 5, 6, 7, 9, 12 and 14 using the following formula: tumor volume (mm3)= L×W2/2, where L and W represent the length and the width of the tumor mass, respectively. The results are shown as the means±standard error (SE) of 3 or 4 mice and were analyzed for statistical significance using a t-test.
Results
Selection of counter ion of HUHS015. To select an adequate counter ion of HUHS015 to improve its solubility in 0.5% MC, we prepared an aqueous solution of HUHS015 at several concentrations (32, 16, 8, and 3.2 mg/ml) with the addition of equimolecular of clinically used counter ions, such as KOH, NaOH or HCl. As a result, the sodium salt of HUHS015 (2) was selected for further studies in this work because the solubility of HUHS015 after the addition of NaOH was increased compared to the other ions (data not shown).
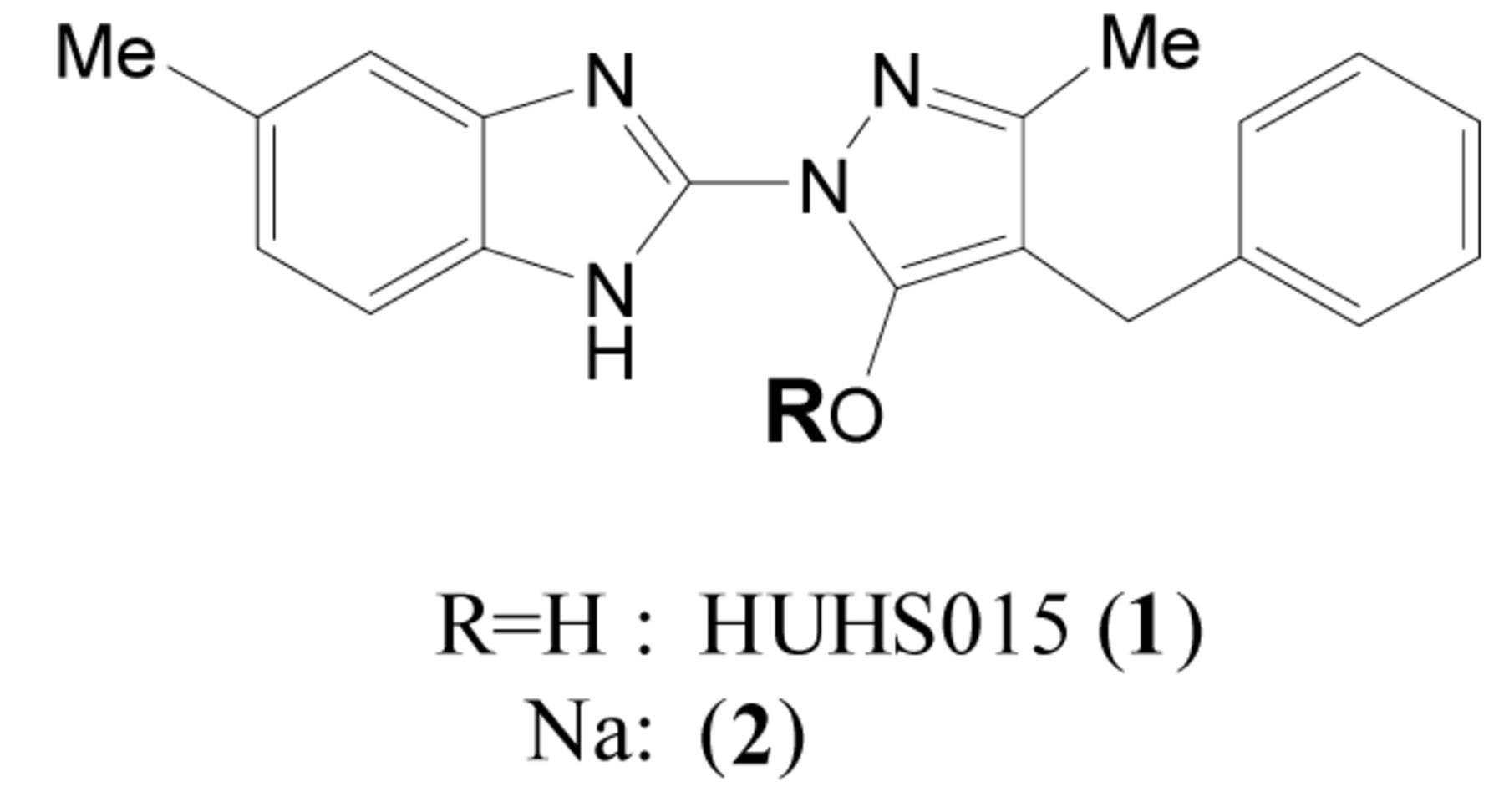
Structure of HUHS015.
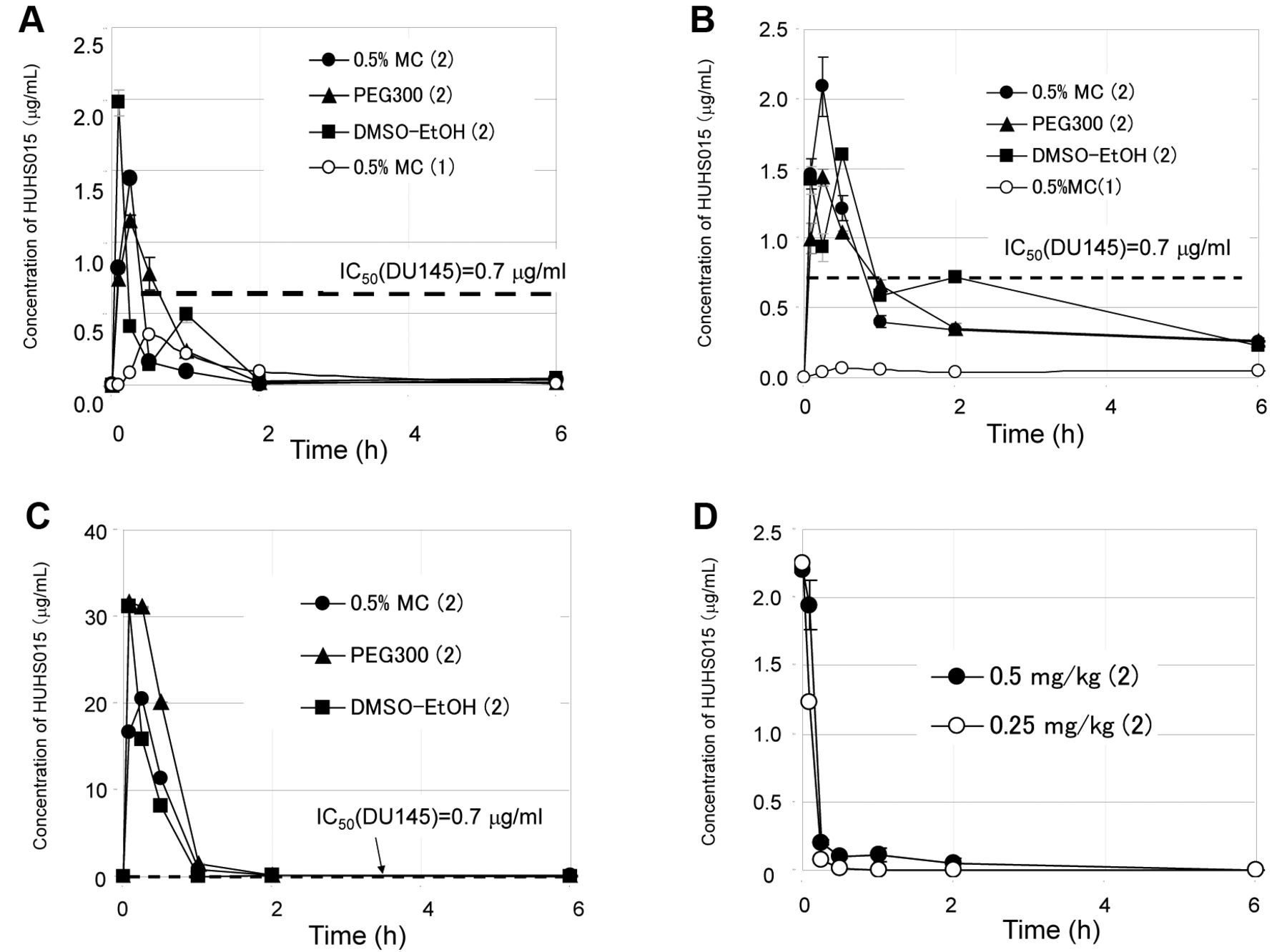
Serum concentration of HUHS015 after oral administration. We first measured the serum concentration of HUHS015 after the oral administration of 1 or 2. HUHS015 (1) was slowly absorbed and reached the maximum concentration (Cmax), 0.35 μg/ml, 30 min after oral administration of 1 in 0.5% MC, as shown in Figure 2A. By contrast, the oral administration of 2 in 0.5% MC resulted in acute absorption and the Cmax value was 1.4 μg/ml at 15 min, which was 4-fold higher than 1. The area under the curve for 24 h after oral administration (AUC0-24) of 2 was 1.4-fold higher than 1 (Table I). We also investigated the effects of solvents for compound administration on the pharmacokinetics of 2. The use of 50% DMSO-15% EtOH or PEG300 as vehicles, resulted in the successful preparation of a clear solution at 32 mg/ml of 2 at room temperature or with heating, respectively (data not shown), while both of them could not dissolve 1. After oral administration of 2 in PEG300, the Cmax of HUHS015 was 1.2 μg/ml at 15 minutes, which was approximately the same for 2 in the 0.5% MC suspension (1.4 μg/ml) (Figure 2A). The AUC0-24 after administration using PEG300 was 1.1 μg • h/ml, which was slightly higher than in 0.5% MC (0.96 μg • h/ml) (Table I). When 50% DMSO-15% EtOH was used as a vehicle, HUHS015 was quickly absorbed and the concentration of HUHS015 reached the maximum at 5 min (Cmax=1.97 μg/ml). Interestingly, the concentration of HUHS015 increased again at 1 h after decreasing once at 30 min (Figure 2A). We now have no information on this phenomenon. The AUC0-24 after the oral administration of 2 in 50% DMSO-15% EtOH was 1.3 μg • h/ml, which was 35% and 88% increased compared to 0.5% MC and 1 in 0.5% MC, respectively. The AUC0-24 values after p.o. administration solution of 2 in PEG300 and 50% DMSO-15% EtOH were improved compared to the suspension of 2 in 0.5% MC. Based on the IC50 value of HUHS015 on the inhibition of DU145 proliferation (0.7 μg/ml), oral administration of 2 enabled to exceed the effective serum concentration in all solvents, whereas 1 did not reach similar serum concentrations.

Concentration of HUHS015 (A) after oral administration (32 mg/kg) of 1 or 2, (B) subcutaneous administration (32 mg/kg) of 1 or 2, (C) intraperitoneal administration (32 mg/kg) of 1 or 2 and (D) intravenous administration of 2 (0.25 or 0.5 mg/kg). A concentration of 32 mg/kg of 1 in 0.5% MC (○) or 2 in 0.5% MC (●), PEG300 (▴) or 50% DMSO/15% EtOH (▪) was orally, subcutaneously or intraperitoneally administered. A concentration of 0.25 mg/kg (○) or 0.5 mg/kg (●) was intravenously administrated with 2 in 50% PEG300. Blood samples were obtained from the abdominal vein after 5, 15, 30 minute, 1, 2, 6 or 24 h. The plasma was separated by centrifugation at 4°C. The concentration of HUHS015 was measured using HPLC after extraction with ethyl acetate from the serum. 1: HUHS015; 2 HUHS015 sodium salt; MC: Methyl cellulose; DMSO: Dimethyl sulfoxide; EtOH: Ethanol; PEG300: Polyethylene glycol 300.
Serum concentration of HUHS015 after subcutaneous administration. The Cmax value after s.c. administration of 2 in 0.5% MC was 2.1 μg/ml at 30 min, which was 33-fold higher than 1 (0.064 μg/ml) (Figure 2B). The AUC0-24 values after s.c. administration of 2 (5.1 μg • h/ml) also improved by 6.7-fold compared to 1 (0.76 μg hr/ml, Table I). The solvent effect on the pharmacokinetics was examined as well. For PEG300, the Cmax was 1.4 μg/ml at 15 minutes (Figure 2B) and the AUC0-24 was 5.1 μg • h/ml, which was the same as 2 in 0.5% MC (Table I). The Cmax after administration of 2 in 50% DMSO-15% EtOH was 1.6 μg/ml (Figure 2B) and the AUC0-24 was 6.0 μg • h/ml, which is 1.2-fold higher than 2 in 0.5% MC (Table I). Therefore, improving the solubility of HUHS015 with 50% DMSO-15% EtOH and PEG300 was also effective for the pharmacokinetics. The use of 50% DMSO-15% EtOH as a vehicle for 2 increased the AUC0-24 value by 40% compared to 0.5% MC and by 7.9-fold compared to the s.c. administration of 1 in 0.5% MC. Importantly, by administering 2 in all solvents, the serum concentration of HUHS015 exceeded the IC50 concentration (0.7 μg/ml) at 1 h, although HUHS015 (1) did not reach this concentration at all.
Pharmacokinetic parameters of 1 and 2 in rats. 1: HUHS015; 2: HUHS015 sodium salt; MC: Methyl cellulose; PEG300: Polyethylene glycol 300; DMSO: Dimethyl sulfoxide; EtOH: Ethanol; p.o.: oral administration; s.c.: subcutaneous administration; i.p.: intraperitoneal administration; i.v.: intravenous administration.
Serum concentration of HUHS015 after intraperitoneal administration. In addition, we measured the serum concentration of HUHS015 after i.p. administration of 2 in 0.5% MC, PEG300 and 50% DMSO-15% EtOH, because i.p. administration is often used in pharmacological tests. In these cases, HUHS015 was acutely and highly absorbed and eliminated in approximately 1 hour (Figure 2C). The Cmax values in 0.5% MC, PEG300 and 50% DMSO-15% EtOH were 20.4, 31.7 and 31.2 μg/ml, respectively, and were more than 10-fold higher compared to p.o. and s.c. and approximately 30-fold higher than the IC50 value (0.7 μg/ml) for DU145 proliferation in vitro. The AUC0-24 values in 0.5% MC, PEG300 and 50% DMSO-15% EtOH were 11.9, 18.6 and 10.0 μg • h/ml, respectively, approximately twice the AUC0-24 after s.c. administration (Table I). The reason why the AUC0-24 value of PEG300 was the highest among them by over 1.5-fold was thought to be the reduced serum volume due to increased peritoneal fluid caused by high osmotic pressure.
Serum concentration of HUHS015 after intravenous administration and calculation of bioavailability. Finally, the intravenous administration of HUHS015 was performed to determine the bioavailability (BA) values. We observed toxic effects of the solvents in the rats for 24 hours at first. Intravenous administration of an aqueous solution of 50% DMSO-15% EtOH caused haematuria, while neat PEG300 caused high osmotic pressure. By contrast, a 50% aqueous solution of PEG300 had a small effect and was selected as the solvent for the i.v. administration. A 0.5 or 0.25 mg/ml solution of 2 in 50% PEG300 was injected and the serum concentration of HUHS015 was determined (Figure 2D). The initial value (C0) was estimated with a free software developed by Kenji Tabata of Fujisawa Pharmaceutical company in Kyoto University, Japan (7). The AUC0-24 after i.v. administration of 2 at 0.25 and 0.5 mg/kg were 0.076 and 0.153 μg • h/ml, respectively (Table I). The linearity between the dosages and AUC0-24 values was confirmed with these two dosages. Therefore, we calculated the BA values of the sodium form of HUHS015 (2), which were 9.8, 11.2 and 13.3% after p.o. administration of 2 in 0.5% MC, PEG300 and 50%DMSO-15%EtOH, respectively, whereas the BA of free HUHS015 (1) was 7.0% (Table I). Therefore, improving the solubility of HUHS015 via the formation of a sodium salt increased, as expected, the BA values.
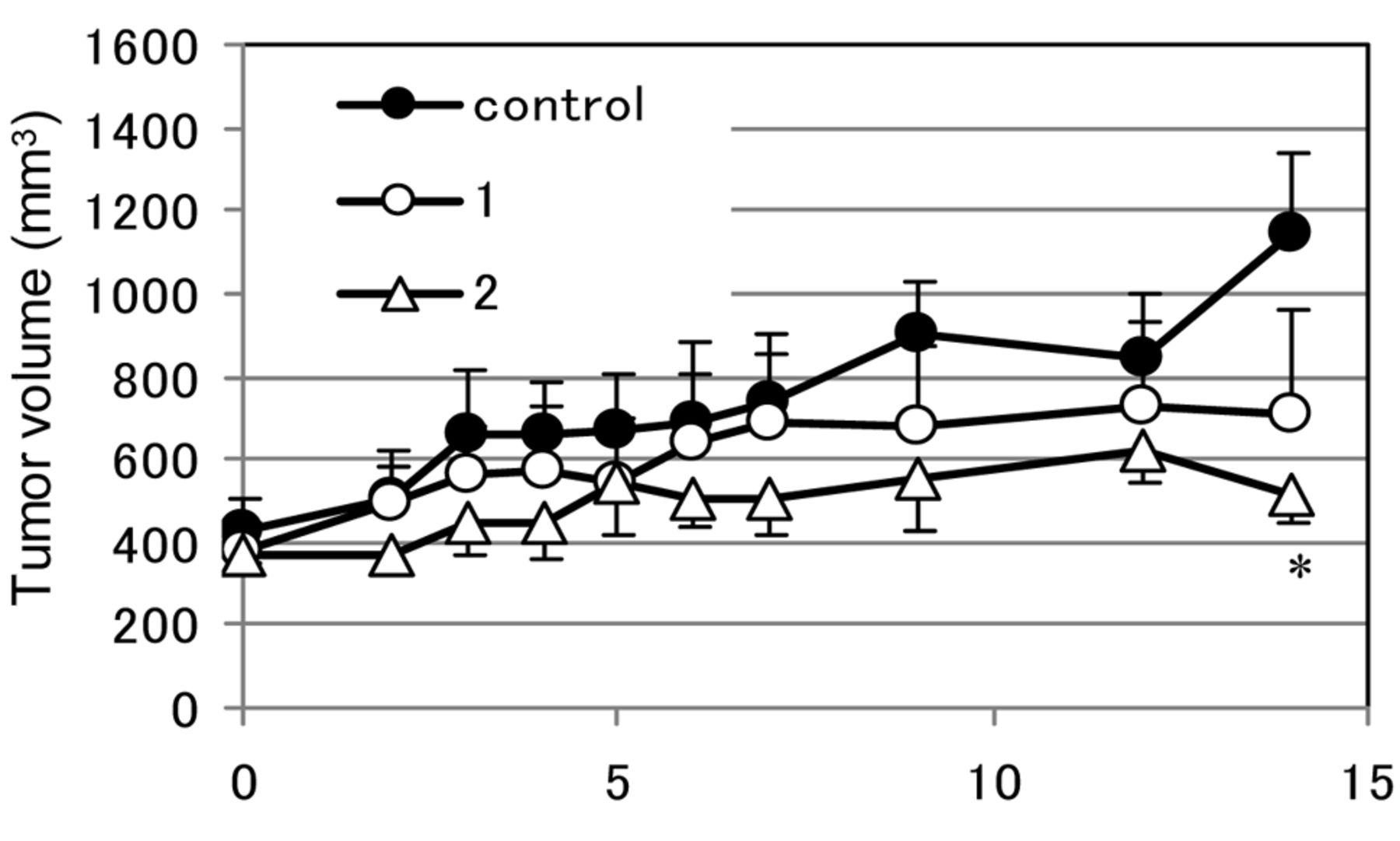
The anti-proliferative effects of 1 and 2 in xenograft mice bearing DU145 tumors (32 mg/kg, s.c.). Mice were subcutaneously administered with 0.5% MC (control), HUHS015 (1) or HUHS015 sodium salt (2) at 32 mg/kg once per day for 13 days. Each point represents the mean±SE of 3 or 4 mice. *p<0.05, comparison with control (Student's t-test). MC: Methyl cellulose; SE: Standard error.
The anti-proliferation effect of 1 and 2 on DU145 xenografts in mice. Finally, we examined the anticancer effect of 2 in a mouse xenograft model. Two weeks after the implantation of DU145 cells with Matrigel™ into nude mice, the mice were divided to 3 groups and subcutaneous administration of vehicle (0.5% MC), 32 mg/kg of 1 or 2 in 0.5% MC was initiated. The administration of 2 significantly inhibited the growth of the tumor after 2 weeks, although that of 1 tended to inhibit the growth of it as well (Figure 3). Insoluble material was observed in the injection area of 1 at the last point, whereas almost no material was detected for 2.
Discussion
We synthezised 1, 3, 4-substituted-1H-pyrazol-5-ol derivatives to identify orally-active small PCA-1 inhibitors and selected 1-(5-methyl-1H-benzimidazol-2-yl)-4-benzyl-3-methylpyrazol-5-ol (HUHS015, 1) for further studies (6). Compound 1 exhibited inhibition of proliferation against DU145 both in vitro and in a xenograft mouse model without observable side-effects after subcutaneous continuous administration for one week (6) and up to one month (data not shown). These results and a previous report of knock-out studies using PCA-1 siRNAs (4) demonstrated that small PCA-1 inhibitors are expected to be effective drugs against prostate cancer without mechanism-based side-effects or toxicity. We have been progressing to prove this concept of PCA-1 inhibitors using HUHS015 (1). However, the inhibitory effect of 1 in vivo is not yet satisfactory. Therefore, we prepared widely used salt formations of HUHS015 in this study and selected the sodium salt of HUHS015 (2) for further studies because of its high solubility in aqueous solutions. Pharmacokinetic parameters of 2 were observed after p.o. and s.c. administration and were compared to 1. The serum concentrations of HUHS015 after both s.c. and p.o. administration of 2 were higher than 1. In particular, s.c. administration of 2 increased the AUC0-24 by 6.7-fold compared to 1. The IC50 value of HUHS015 on DU145 cell proliferation in vitro was 0.7 μg/ml and administration of 2 succeeded in exceeding this IC50 value, whereas 1 did not. We also observed a solvent effect on the absorption of 2. PEG300 and 50% DMSO-15% EtOH were used as a solvent compared to a standard 0.5% MC suspension. Among them, 50% DMSO-15% EtOH was the most effective solvent and was easy to prepare an aqueous solution of 32 mg/ml HUHS015 at room temperature, although the PEG300 preparation required heating. As a result, both 50% DMSO-15% EtOH and PEG300 improved the pharmacokinetics of 2 after p.o. and s.c. administration and we succeeded in increasing the AUC0-24 by 7.9-fold compared to 1 in 0.5% MC, as investigated in previous studies (6).
Finally, we compared the anti-cancer effects of 1 and 2 in a xenograft model (Figure 3). Compound 1 or 2 in 0.5% MC was administered subcutaneously once per day for 13 days to nude mice bearing a DU145 mass and both inhibited the growth of the DU145 tumour mass. Administration of 2 significantly inhibited the growth of DU145 cells and was more effective than 1. There were lumps of undissolved HUHS015 at the injection area of 1, although there were almost no lumps for 2. Therefore, improving the solubility of HUHS015 with a sodium salt formation increased the BA (Table I) and anticancer effects of HUHS015 in this study, as expected. We postulated that the relatively potent inhibitory effect of 1 in the xenograft model (Figure 3) compared to the low serum concentration, which was almost 1/9 of the IC50 (Figure 2B), was due to diffusion effects of HUHS015 by subcutaneous infiltration. We are continuing our studies of the anticancer effects of HUHS015 (1) or its sodium salt (2) in vitro and in vivo using single or combination therapy, as well as the design and synthesis of novel derivatives of HUHS015.
Acknowledgements
This research was supported in part by a Grant-in-Aid for Scientific Research (C) provided by the Program for Promotion of Fundamental Sciences in Health Sciences of the National Institute of Biomedical Innovation (NIBIO) and by a Grant-in-Aid from the Knowledge Cluster Initiative (Second Stage) of the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
- Received September 29, 2014.
- Revision received October 22, 2014.
- Accepted October 27, 2014.
- Copyright © 2015 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.