


Abstract
Cytochrome P450 2E1 (CYP2E1) has active roles in bioconversion and biotransformation in humans. Although predominantly present in hepatocytes, CYP2E1 has also been found in hematopoietic stem cells and subtypes of acute myeloid leukemia with unknown clinical significance except for the metabolism of anti-fungal drugs. In the present study, we demonstrated a novel role of CYP2E1 inducing megakaryocyte development in human hematopoietic stem cells and leukemia bipotent K562 cells. CYP2E1 was induced by phorbol-12-myristate-13-acetate in dose-dependent manner in K562 cells as well as in hematopoietic stem cells by thrombopoietin, and ingenol 3,20-dibenzoate (IDB), respectively. Overexpression of CYP2E1 was positively correlated with megakaryocytes and in megakaryocyte maturation. In addition, plasmid-driven expression of CYP2E1 in K562 cells led to morphological transformation of leukemic blasts to pro- and mature megakaryocytes. In contrast, knockout of CYP2E1 by specific interfering RNA diverted these cells to erythroid differentiation. Finally, treatment of K562 cells by a free radical scavenger, N-acetyl L-lysine significantly inhibited CYP2E1 and megakaryocyte differentiation. In summary, our data demonstrated that activation of CYP2E1 and reactive oxygen species signaling promotes megakaryocyte development.
Megakaryocytopoiesis is a cellular developmental process prior to the release of platelets into the circulation. Regulation of megakaryocytopoiesis is a complex process that begins with commitment of hematopoietic stem cells to maturation into progenitor cells regulated by a number of growth factors (1). The binding of thrombopoietin to its receptor, a human homolog of the murine myeloproliferative leukemia virus (c-MPL), on cluster differential antigen 34 (CD34)-positive cells initiates the activation of signal cascades, including mitogen activated protein kinase or extracellular signal-regulated kinases (MAPK/ERK), p38MAPKs, and and Jun Proto-oncogen (c-JUN), which leads to megakaryocyte growth and differentiation, as well as platelet production (2-5). It has long been recognized that protein kinase C (PKC) agonists, such as phorbol esters, can promote megakaryocyte differentiation of human leukemia cell lines with the potential to undergo megakaryocyte differentiation (6). One such cell line is erythroleukemia K562. K562 cells have been extensively studied as a model for megakaryocyte differentiation due to their biphenotypic (erythroid/megakaryocytic) nature (7-9). Previous studies have demonstrated that MAPK/ERK activation plays a role in PKC-induced megakaryopoiesis, but how this is maintained is as yet unknown (10-12).
Recently, studies on the role of reactive oxygen species (ROS), such as superoxide anion (O2−), hydrogen peroxide (H2O2) and hydroxyl radical (OH•) in regulation of hematopoeisis and megakaryocyte differentiation have been reported (13-15). It was demonstrated that in response to hematopoietic growth factors, ROS might activate tyrosine phosphorylation of growth factor receptors such as the early growth response protein 1(EGR1) expression during hematopoietic differentiation (16). Sources of ROS under normal conditions in humans might originate from the leakage from NADP oxidase during oxidative phosphorylation, multiple cytoplasmic xanthine, NADPH oxidases, and cytochrome P450 (17, 18).
Cytochrome P450 2E1 (CYP2E1), a member of the cytochrome P450 mixed-function mono-oxygenase system, is well-known for its active roles in bio-conversion and biotransformation, and generation of ROS with or without substrate (18-22). In vitro-reconstituted CYP2E1 in intact cells by transient transfection has also been shown to produce both O2− and H2O2 (19-22). Although its major location is the liver, CYP2E1 is overexpressed in acute myeloid leukemia with t(8;21) and inv(16), as well as inducibly in K562 cell, an erythroleukemia cell line (31).
Primers used in this article.
Understanding these mechanisms may provide insight into the CYP2E1 and oxidant signaling components as potential therapeutic targets. To address this issue, we investigated the effects on megakaryocytic differentiation of up- or down-regulation of expression of CYP2E1 in K562 cells by phorbol-12-myristate-13-acetate (PMA), as well as in human CD34+ progenitor cells induced by thrombopoietin, erythropoietin, and ingenol 3,20-dibenzoate (IDB) and their correlations with expressions of differentiation related markers such as CD9, CD41 and EGR1 (23-24).
Materials and Methods
Reagents. All cell culture plates, media, fetal bovine serum (FBS), antibiotics (penicillin and streptomycin), and culture supplements were obtained from Life Technologies (Grand Island, NY, USA) unless otherwise specified. Nucleofector Kit V was purchased from Amaxa (Cologne, Germany). K562 cell line (CCL-243TM) was purchased from the American Tissue and Cell Culture Collection (ATCC, Manassas, VA, USA). The CellTiter 96® Aqueous One solution cell proliferation assay (MTS) kit was purchased from Promega (Madison, Wl, USA). N-Acetyl-L-lysine (NAC) and PMA were purchased from Sigma (St. Louis, MO, USA). U0126, an ERK inhibitor, was obtained from Cell Signaling (Beverly, MA, USA). Dimethyl sulfoxide (DMSO), Geneticin (G418), recombinant human stem cell factor (rhSCF), RNA stabilizer Trizol, Superscript reverse transcriptase (RT) reaction kit, and Taq DNA polymerase were obtained from Invitrogen (Carlsbad, CA, USA). The cloning pcDNA3.1 (pVEC) and expression pcDNA3.1-CYP2E1 plasmids (p2E1) were generously provided by Dr. Cederbaum at Mount Sinai School of Medicine, NY, USA, and were proven to express a functional protein (22). SYBR Green PCR master mix was purchased from Life Technologies (Grand Island, NY, USA).
Culture of K562 cells. K562 cells were grown in complete medium (CM) (Iscove's modified Dulbecco's medium supplemented with 10% FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 2 mM L-glutamine) at 5×105 cells/ml. The cells were routinely seeded at a density of 2×105/ml and cultured at 37°C in 5%CO2 and the medium was refreshed every 2 days.
Cell treatment assay with PMA, NAC and U0126. K562 cells (2×105 cells/well) were cultured with different doses of PMA (1-100 nM) for 48 h. For treatment with NAC, K562 cells (1×106/ml) were treated with 20 μM NAC for 30 min, washed three times with FBS-free CM, and resuspended in CM and cultured for 24 h. For U0126 treatment, K562 cells (1×106/ml) were treated with 10 μM U0126 (Cell Signaling) for 2 h, and PMA (10 nM) was then added and cells cultured for an additional 24 h. DMSO was used as mock treatment for all experiments. Cells were harvested followed by three washes with 1× PBS. Cell pellets were then processed for RNA or protein isolation.
Human CD34+ stem cell culture and treatment with erythropoietin, thrombopoietin, or IDB. The human CD34+ hematopoietic progenitor cells were cultured and treated as previously described (24). Briefly, CD34+ cells (1×106/vial) were cultured with StemSpan SFEM medium (StemCell Technologies, Vancouver, Canada) with 25 ng/ml of rhSCF for 48 h at 37°C in 5% CO2 (24). For induction of erythroid or megakaryocyte differentiation, vehicle (DMSO), or 0.5 U/ml erythropoietin, or 40 ng/ml thrombopoietin were added in SFEM medium and cells were cultured for 12 days. For induction by 10 nM IDB, the cells were harvested at 48 h after the addition.
Establishment of stable pVEC/K562 and p2E1/K562 cells. K562 cells (2×106) at log phase (approximately 24 h of culture after seeding) were transfected with 1.0 pg pcDNA3.1-CYP2E1 (p2E1/K562) or pcDNA3.1 DNA (pVEC/K562) by nucleofection (Amaxa) according to the manufacturer's instructions. Immediately afterwards, cells were diluted in 2 ml CM to 5×105 cells/ml and cultured at 37°C in 5% CO2. Forty-eight hours after transfection, cells were washed and serially diluted to a low cell density from 10-100 cells/ml, and 100-μI aliquots were plated onto 96-well plates in the presence of 1 mg/ml geneticin and cultured for 21 days. Individual G418-resistant colonies were selected, expanded and tested for integrated DNA and expression of CYP2E1. Genomic DNA (gDNA) was isolated and the presence of CYP2E1 cDNA inserts was verified in established p2E1/K562 cells but absent from pVec/K562 cells by PCR using primers flanking vector-CYP2E1 or vector-vector sequences (Table I). After verification, the established cells underwent single-cell cloning by serial dilution and cells were stored in liquid nitrogen for future use. These cell lines were maintained under the same normal growth conditions as the parental K562 cells without G418.
Knockdown of CYP2E1 by siRNA. The expression of CYP2E1 was silenced by nucleofection with small interfering RNA (siRNA) specific for CYP2E1-encoding sequences (2E1-siRNA) or non-target random sequences (RS-siRNA) (ThermoFisherScientific, Dharmacon, MA, USA) at 3 pg/106 cells. After 24 h, the transfected cells were treated and the cells were harvested for further analysis.
DNA and RNA isolation, cDNA reverse-transcription, PCR and Quantitative real-time PCR (qRT-PCR). gDNA was isolated from p2E1/K562 cells using QIAam DNA Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions and dissolved in Tris-EDTA (TE) buffer. The PCR reaction was carried out in a volume of 25 μI with HotStarTaq Master Mix Kit (Qiagen) containing 10 ng of genomic DNA and 0.5 μM primers (Table I). The amplified DNA fragments encoding pCYP2E1 or control pCND3.1 were detected by electrophoresis on 2% agarose gel.
RNA was isolated from K562 cells, and established p2E1/K562 or pVEC/K562 cells using TRIzol® Plus RNA Purification Kit according to the manufacturer's procedure (Invitrogen) followed by cDNA reverse transcription by using Superscript™ II Reverse Transcriptase with 1 pg RNA according to the manufacturer's procedure (Life Technologies). End-point PCR for the expression of target genes was performed as described previously (23). The primers for amplification of the tetraspanin family (CD9), platelet glycoprotein IIb (CD41), EGR1, glycophorin A, CYP2E1 (2E1), plasmid CYP2E1 (p2E1) and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) are listed in Table I. The amplicons detected by electrophoresis on 2% agarose gel were visualized under UV light and images were taken by an alpha-image system (alpha-image). The quantitative RT-PCR was performed with a QuantiTect RT Kit (Qiagen) according to manufacturer's instructions. The qRT-PCR was performed as described using SYBR Green PCR master mix, purchased from Life Technologies (Grand Island, NY, USA) and utilized on an Applied Biosystems 7500 Fast Real-time system (38). Fold changes were used for expression differences between treated and control groups, and calculated as described by Pfaffl (39).
Western blot analysis. The expression of proteins were determined by western blot analyses as described previously (34). Briefly, K562 cells (1×106) were harvested, washed with PBS and resuspended in 1×Laemmli sample buffer containing proteinase and phosphatase inhibitors. Protein concentration was determined using Bio-Rad protein assay. Fifty picograms of proteins were separated on sodium dodecyl sulphate polyacrylamide gel electrophoresis and electrophoretically transferred to polyvinylidene difluoride membranes. Membranes were probed with antibodies against phosphorylated pERK1/2 (Cell Signaling, Beverly, MA, USA), EGR1, CD9, CD41, glycophorin A (Santa Cruz Biotechnology, Dallas, TX, USA), and CYP2E1 (Chemicon, Temecula, CA, USA). The blots were immersed in ECL reagent (Amersham, Arlington Heights, IL, USA) and then exposed to autoradiography film (Denville, Metuchen, NJ, USA). The blots were visualized with ECL reagent (Amersham) and then exposed to Hyperfilm (Amersham). The densities of each band on the blot were quantified by Photoshop (Adobe Systems Inc., San Jose, CA, USA) and expressed as fold-change compared to the controls.
Morphology study. Cell morphology of established p2E1/K562 and pVEC/K562 cells was visualized using an inverted microscope (Olympus CKX31; Olympus, Center Valley, PA, USA) captured at ×40 magnification with a digital camera (C-5060; 0.13 aperture; Olympus). Total 1×104 viable cells were spread by centrifugation (Shandon Cytospin 3; Thermo Fisher, Waltham, MA, USA) on slides, and fixed with ice-cold acetone, and stained by Diff-Quick (LabAids Inc., Ronkonkoma, NY, USA) or Wright staining (Sigma). At least 200 cells were examined for megakaryocytic differentiation microscopically.
Statistical analysis. A two-tailed paired t-test used to compare the statistical significance of differences in data from the two groups. In the design of the experiments, triplets were repeated at least three times and results are expressed as means and standard deviation (SD).
Results
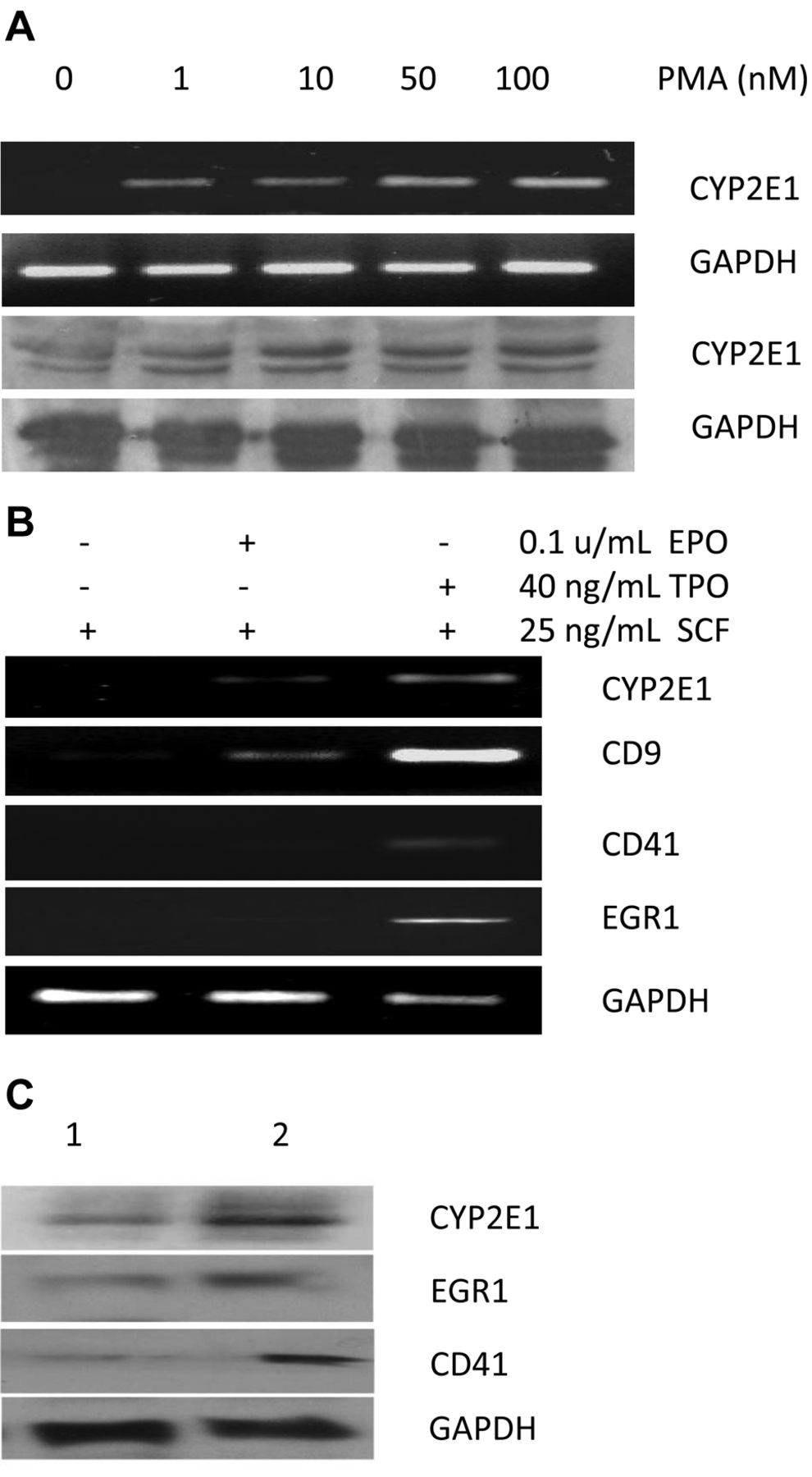
Correlation between CYP2E1 expression and megakaryocyte differentiation. Dose-dependent and time-dependent CYP2E1 induction was performed on K562 cells induced by PMA. Both mRNA and protein expression of CYP2E1 in K562 cells was induced by 1 to 100 nM PMA (Figure 1A) and CYP2E1 was detectable at 24 h and sustainably expressed up to 72 h (data not shown). To determine whether CYP2E1 is also inducible in physiological hematopoiesis, measurement was performed at day 12 for human CD34+ progenitor cells post-treatment with erythropoietin, thrombopoietin, or IDB (24). As shown in Figure 1B, CYP2E1 RNA was not present in day 12 culture in 25 ng/ml SCF (Lane 1, Figure 1B), minimally induced by 0.5 U/ml erythropoietin (Lane 2, Figure 1B), but remarkably induced by 40 ng/ml thrombopoietin (Lane 3, Figure 1B). The levels of CD9, CD41 and EGR1 have similar patterns to that of CYP2E1 under these conditions (Lanes 1-3, Figure 1B). IDB is a novel PKC isoform agonist to enhance megakaryopoiesis in cultured primary CD34+ progenitor cells (23-24). In the presence of 100 nM IDB, CYP2E1, CD9 and CD41 proteins were up-regulated during IDB-induced megakaryocyte differentiation at 48 h (Lane 2, Figure 1C) compared to cells treated with DMSO (Lane 1, Figure 1C). The concurrent expression of CYP2E1 in human CD34+ progenitor cells with megakaryocyte differentiation markers further suggests the positively-regulating role of CYP2E1 in this event.
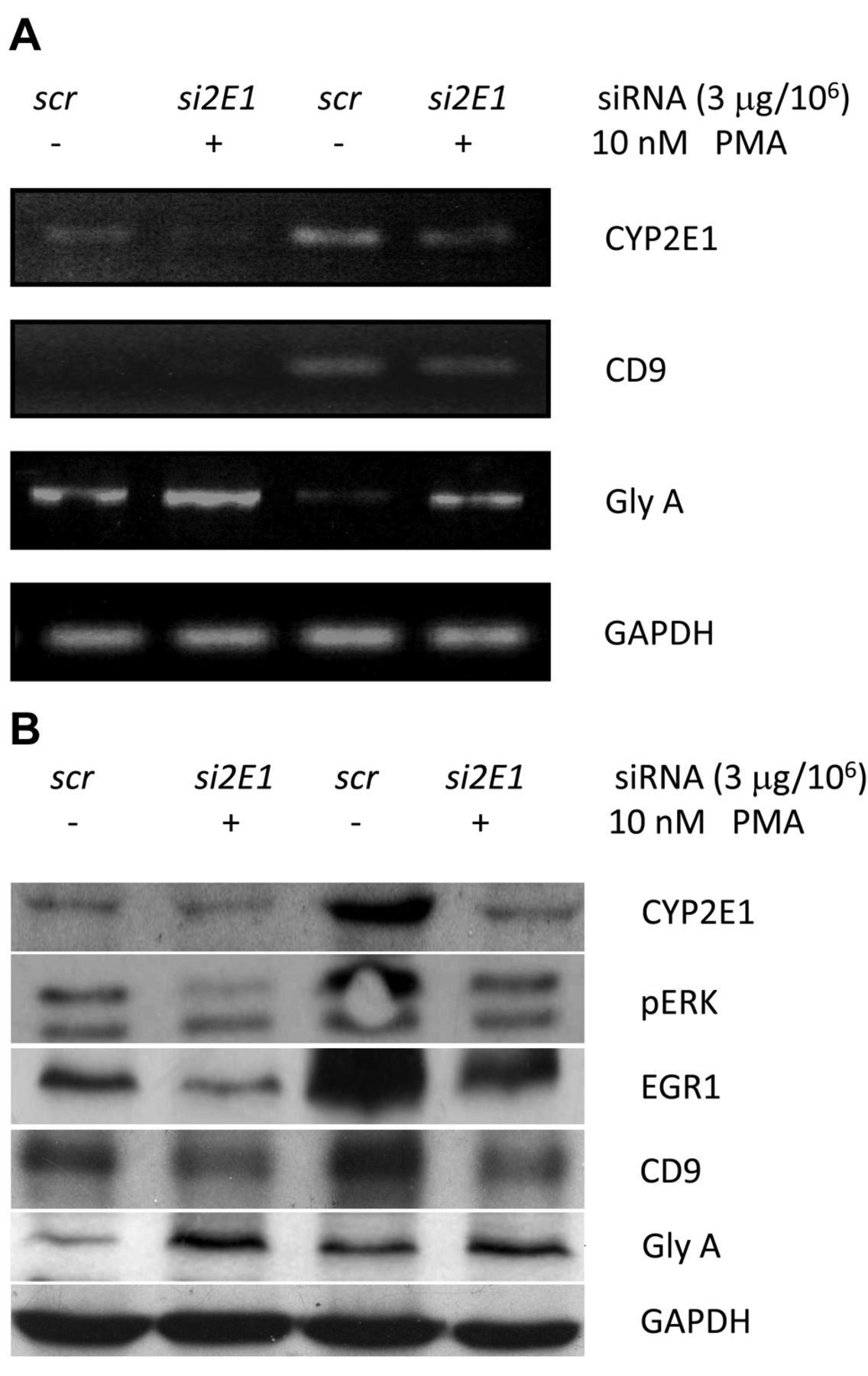
Down-regulation of CYP2E1 suppressed megakaryocyte differentiation. We next examined whether knockdown of the expression of CYP2E1 by its specific small interference RNA (2E1-siRNA) would affect PMA-activated megakaryocyte differentiation. As shown in Figure 2, both basal expression and PMA-induced CYP2E1 RNA and proteins were suppressed by target-specific 2E1-siRNA (Lanes 2 and 4, Figure 2) compared to those treated by the sham control siRNA (Lanes 1 and 3, Figure 2). The RNA and protein expression of CD9, pERK and EGR1 were also suppressed (Lanes 2 and 4, Figure 2) compared to the basal expressions (Lanes 1 and 3, Figure 2). In contrast, silencing CYP2E1 by 2E1-siRNA remarkably enhanced expression of glycophorin A, a sialoglycoprotein solely present on human erythrocyte membranes and a marker of erythroid differentiation and maturation (Figure 2) whether PMA was present or not.
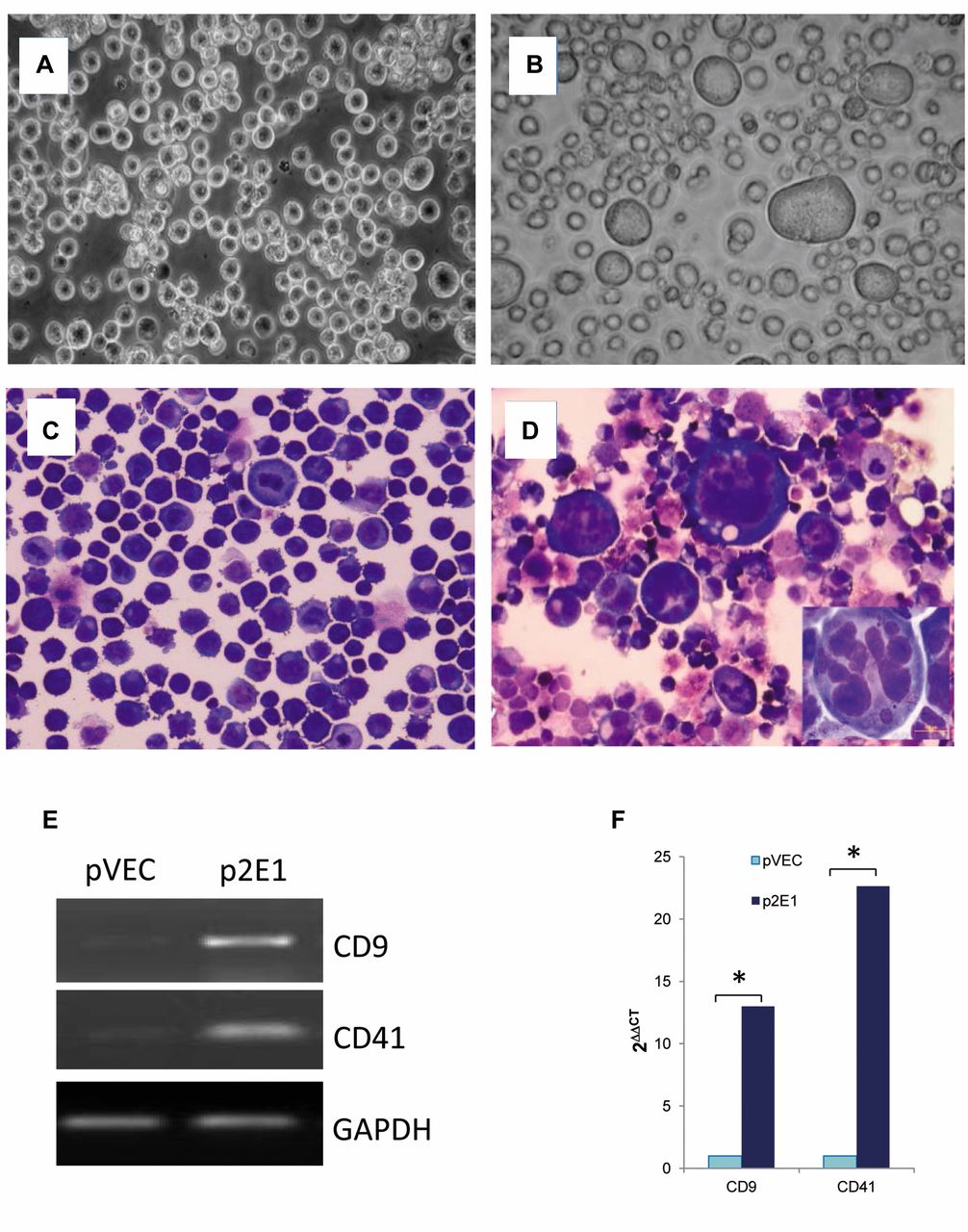
Stably-expressed CYP2E1 promotes morphological and phenotypic differentiation of K562 cells to megakaryocytes. Next, K562 cells were transfected with CYP2E1 expression plasmid, pcDNA3.1-CYP2E1, or vector, pcDNA3.1. The stable cells were established by G418 selection (see Materials and Methods), and designated as p2E1/K562 and pVEC/K562, respectively. The chromosomal integrations of both plasmids were examined by PCR amplification of the gDNA extracted from the established cell lines, p2E1/K652 and pVEC/K562, respectively, using primers designated to amplify only integrated pcDNA3.1 sequences or cDNA sequences of CYP2E1 (Table I). This approach confirmed that PCR products were from the integrated gDNA but not from the endogenous cDNA of CYP2E1. Morphologically, there were more large-(29.0±3.6% vs. 3±1.7%, p<0.001) and medium- (62±3.6% vs. 2.3±2.1%, p<0.001) sized p2E1/K562 cells with lobulated nuclei than pVEC/K562 cells (Figure 3). Pseudopodia, however, were not seen in both established cell lines. Conventional and real-time PCR were performed and confirmed the overexpression of both CD9 and CD41 in p2E1/K562 cells than in pVEC/K562, respectively (12.99- and 22.63-fold, p<0.001). Meanwhile, overexpression of CYP2E1 was confirmed both at RNA and protein levels in p2E1/K562 cells compared pVEC/K562 cells under conventional culture conditions.

Induction of CYP2E1 expression in human K562 erythroleukemia cells and CD34+ progenitors cultured with TPO, EPO or IDB. A: K562 cells (2×105 cells/well) cultured with different doses of or without PMA as indicated for 48 h. Cells were harvested for preparation of RNA and protein as described in the Materials and Methods. CYP2E1 RNA (upper two panels) and protein (lower two panels) expressions were examined by 2% agarose gel electrophoresis and immunoblot, respectively. B: Human CD34+ hematopoietic progenitor cells were cultured with DMSO (Lane 1), or 1.0 U/ml EPO (Lane 2), or 40 ng/ml TPO (Lane 3) for 12 days. Total RNA was extracted, and expression of CYP2E1, CD9, CD41 and EGR1 were examined by real-time polymerase chain reaction. C: In the same manner as in B, CD34+ cells were cultured with DMSO (Lane 1), or 100 nM IDB (Lane 2) for two days. The expressions of CYP2E1, CD9, and CD41 were examined by immunoblot. GAPDH expression was used as an internal control for all tests.
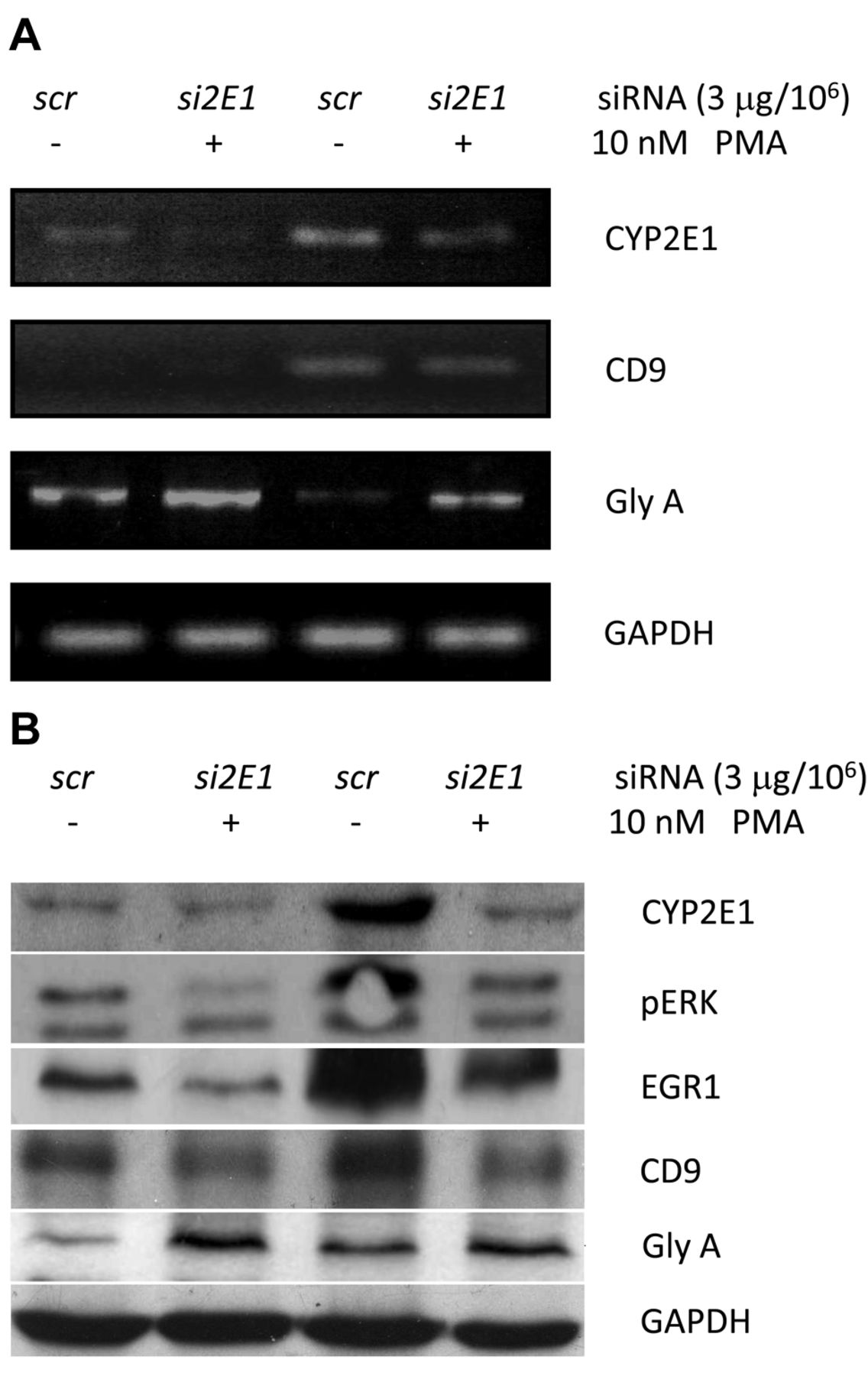
Silencing of CYP2E1 expression in K562 cells suppressed megakaryocyte differentiation. CYP2E1 expression was knocked-down specifically by siCYP2E1 at 3 μg/106 cells and then cells were cultured with or without 10 nM PMA (as described in the Materials and Methods). RNA levels of CYP2E1, CD9, glycophorin A (GlyA) were examined by real-time polymerase chain reaction (A) and protein levels of CYP2E1, phosphorylated ERK (pERK), ERG1, CD9, and GlyA were examined by immunoblot (B). Lane 1: Control with sham siRNA (so) and DMSO; lane 2: specific siCYP2E1 and DMSO; lane 3: sham siRNA (scr) with 10 nM PMA for 48 h; and lane 4: siCYP2E1 with 10 nM PMA for 48 h. GAPDH expression was used as an internal control for all tests.

Constitutively expressed CYP2E1 in p2E1/K562 cells enhances megakaryocyte differentiation. Phase-contrast microscopy (A,B) and Quick-Diff stain (C, D) show increased numbers of large cells with multinuclei or increased lobation of nuclei in p2E1/K562 cells (B and D) but less often in pVEC/K562 cells (A and C). Expressions of CD9 and CD41 in pVEC/K562 and p2E1/K562 cells were examined by conventional Real-time polymerase chain reaction (E) and quantitative real-time PCR (F). The data are presented as the mean of triplets in three independent tests, *p<0.001.

Diminution of ROS and ERK expression blocks megakaryocyte differentiation. Effects of ROS (A) and ERK (B) on CYP2E1 expression and megakaryocyte differentiation was studied in K562 cells treated with 20 μM N-acetylcysteine (NAC) or 10 μM ERK inhibitor U0126 followed by 10 nM PMA. Immunoblotting for CYP2E1, CD9, phospho-ERK, and EGR1 was carried-out 48 h later after treatment with PMA.
Treatment of K562 cells with NAC or ERK inhibitor, U0126, blocks megakaryocyte differentiation. A previous study has shown that intracellular ROS affect hematopoiesis. Since CYP2E1 is an active producer of ROS, we postulated that CYP2E1 might induce megakaryocyte differentiation via production of ROS which then activates the MAPK/ERK signaling pathway. K562 cells were first treated with 20 μM NAC without or with 10 nM PMA. Expression of CYP2E1, CD9, and pERK was up-regulated in the presence of 10 nM PMA (Lane 2, Figure 4A), but significantly reduced when cells were treated with 20 μM NAC (Lane 3, Figure 4A). Similarly, pERK, EGR1, and CYP2E1 expressions were remarkably up-regulated by 10 nM PMA (Lane 3, Figure 4B), but dramatically suppressed when U0126, an MAPK/ERK kinase inhibitor, was present (Lane 4, Figure 4B).
Discussion
Human megakaryocytopoiesis is the hematopoietic differentiation process that depends on the signaling provided by growth factors such as thrombopoietin to their cognate receptors on hematopoietic progenitor cells, followed by the activation of the MAPK signal pathway(s), which results in the transcription of genes critical for the onset of differentiation. Thrombopoietin-induced in vitro megakaryocytic differentiation from CD34+ stem cells is a lengthy process and may take several days to accomplish. To maintain the process of maturation, the intracellular levels of EGR1, a C2H2 type zinc-finger nuclear protein important in controlling mitogenesis and differentiation, are critical for this process (12, 25). In this study, we uncovered a novel role of CYP2E1 in megakaryopoiesis through its intermediate but specific role in activation of MAPK/ERK pathway, possibly via ROS activation, leading to the induction of EGR1, CD9, and CD41 expression in megakaryocyte differentiation.
Although polymorphisms of CYP2E1 have been reported by others to be linked to an increased risk for malignancy (26), and the increased expression of CYP2E1 has been reported in association with ethanol-induced liver disease due mostly to post-transcriptional CYP2E1-dependent oxidative stress (22), little is known about its role in hematopoietic differentiation. Elevated expression of CYP2E1 was observed in two types of acute myeloid leukemia with recurrent gene re-arrangements, t(8;21)(q22;q22) and inv(16)(p13q22)/t(16;16)(p13;q22), but cause-effect studies were not performed (27, 30). Recently, the implications of pathogenesis in acute myeloid leukemia with inv(16) was revealed (31). The functional role of overexpression of CYP2E in hematopoiesis has not been reported.
In the present study, we demonstrated that CYP2E1 was induced and overexpressed in PMA-stimulated K562 cells, and IDB and thrombopoietin induced CD34+ progenitors, along with the co-expression of EGR1, CD9 and CD41. Knockdown of CYP2E1 by specific siRNA resulted in suppression of EGR1 and megakaryocyte-specific markers such as CD9 and CD41 (25, 33, 35). In contrast, K562 cells switched to erythroid differentiation when CYP2E1 was specifically knocked-down. Established cells with constitutive expression of CYP2E1 promoted K562 cell differentiation toward megakaryocytes morphologically and phenotypically. These results, for the first time, explored the novel rule of CYP2E1 for megakaryocyte differentiation in a specific but regulatory manner.
The precise mechanism of CYP2E1-mediated megakaryocyte differentiation is unknown but involvement of ROS is highly suggested. Study by Besancenot et al. showed that thrombopoietin can induce cell-cycle arrest in the megakaryocyte UT7-MPL cell line by the activation of the MAPK/ERK pathway, induction of EGR1 transcription, and ROS production (34-35). However, the sources of ROS production are still under debate (13, 17, 19, 25, 36). As an effective generator of ROS, CYP2E1 produces powerful oxidants such as hydroxyl radicals in the presence of ethanol and might cause DNA damage (20, 22). Studies on Cyp2e1-knockout mice revealed reduced cytotoxicity in ethanol-induced hepatic fibrosis, as well as oxidative stress (33). We demonstrated in this study that elimination of ROS in K562 cells by the free-radical scavenger NAC mimicked the results of silencing of CYP2E1 expression by siRNA, and therefore, suggest that ROS might be downstream of CYP2E1 in the regulation of megakaryocyte differentiation.
Furthermore, we demonstrated that MAPK/ERK inhibition partially blocks CYP2E1. This result would suggest a re-enforced signaling circuit among CYP2E1/ROS and ERK/EGR1 to form a positive feedback loop to turn on the switch for megakaryocyte differentiation. This loop might also be the driving factor for sustained activation of MAPK/ERK signals in prolonged megakaryocyte differentiation and maturation, given the fact that CYP2E1 was also actively induced by thrombopoietin in day 12 cultures of human CD34+ progenitor cells but not in erythropoietin-treated cultures (Figure 1B). The signalling pathway and the re-enforcing loop of activation of CYP2E1-mediated megakaryocyte differentiation might exist but further studies are needed to confirm this.
Whether or not CYP2E1 may have transcriptional factor activity has not been revealed. It is very interesting that while searching by two-hybrid analysis for proteins that associate with the Fanconi anemia group G protein (FANCG), Futaki et al. identified a novel interaction between FANCG and CYP2E1 which might alter redox metabolism and increase DNA oxidation in mammalian cells (37). Our results show that knockdown of CYP2E1 expression in K562 cells resulted in direct inhibition of MAPK/ERK, EGR1, CD9, and CD41, and enhanced expression of glycophorin A, which may suggest its potential involvement in gene regulation either directly or indirectly by binding to DNA-binding proteins, including transcriptional factors. Further studies are required to support the latter hypothesis.
Conclusion
In summary, lineage differentiation is precisely regulated to ensure morphogenesis, maturation, and development. Among the signaling and differentiation factor networks, erythropoietin, thrombopoietin, MAPK/ERK, and even free radicals, interact to ensure megakaryocytic/erythroid differentiation. Our findings demonstrate that CYP2E1 specifically acts at the erythroid-megakaryocytic differentiation bifurcation and favors megakaryocyte differentiation when overexpressed in human progenitor cells.
Acknowledgements
The Authors acknowledge administrative assistance of Shelly Belcher, and Amy Glaze from Department of Pathology, The Ohio State University, Columbus, OH. This work was partially supported by American Cancer Society Grant # IRG-67-003-47 to WZ.
Footnotes
-
Conflicts of Interest
The Authors have no conflicts of interest.
- Received June 23, 2014.
- Revision received July 23, 2014.
- Accepted July 25, 2014.
- Copyright © 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved




{kind=link}
{kind=link}
{kind=link}
{kind=link}