


Abstract
The in vivo absorption of didanosine was studied, focusing on the performance of a novel pharmaceutical formulation for didanosine, composed of chitosan granules containing didanosine incorporated in chitosan microspheres. This novel formulation is aimed at oral administration in AIDS therapy. The experimental results in male adult dogs showed controlled delivery of didanosine along 36 h, with a 2-fold increase in the absorption time of didanosine compared to the commercial granules, gastro-resistant didanosine and tablets. The higher absorption is due to adhesion to the intestinal membrane, improving absorption through increase of residence time, permeation and release. Furthermore, the novel formulation facilitates handling and deglutition, especially in the elderly and children, as well as enhances the taste and reduces the frequency of doses and collateral effects associated with a high concentration of the buffer agents usually used in other formulations.
Didanosine is an important drug used in AIDS therapy (11, 12). It is a nucleoside analog of adenosine which prevents replication of human immunodeficiency virus (HIV) by inhibiting HIV reverse transcriptase (4). Didanosine is a component of highly active anti-retroviral drug combination therapies (2, 4). It is also particularly used for zidovudine-resistant patients (25).
In Brazil, free didanosine is usually administered in buffered tablets to prevent drug de-acetylation when exposed to the low pH of the stomach. The tablet formulation requires the addition of 50% of carbonate or magnesium hydroxide to reach the buffering effect. However, some collateral effects, such as diarrhea and renal problems, are associated with such agents at such concentrations. Furthermore, the buffered tablets are large and hard to swallow (1). Great benefits in didanosine administration were obtained in 2001 with the commercial formulation of gastro-resistant granules, Videx® EC (Bristol-Myers Squibb), but the cost was reported to be high, which remained unfeasible regarding its distribution by the government agencies in developing countries like Brazil (13).
Advances in polymer and colloid sciences, as well as in micro- and nanotechnologies for drug encapsulation, have generated novel pharmaceuticals with enhanced pharmacological properties (26). Chitosan is a natural polymer obtained from deacetylation of chitin, which is abundant in the exoskeleton of crustaceans (9). Due to its abundance, low price, as well as its physicochemical properties, chitosan has been used as a bioactive or biomaterial in pharmaceutical and cosmetic applications (6, 14, 15)
We recently developed chitosan mucoadhesive granules containing chitosan microspheres crosslinked with sodium tripolyphosphate (TPP) for drug encapsulation by ionotropic gelation (17, 19). Didanosine was efficiently incorporated into the microspheres which were formulated as granules prepared by extrusion and spheronization. This novel formulation was stable, reduced the concentration of calcium carbonate buffer required to prevent the deacetylation of didanosine under acidic pH and promoted controlled release of didanosine. We also studied the in vitro permeation of didanosine from these granules across the three different segments of rat intestine using the everted gut sac model. The performance of didanosine permeation from the chitosan granules containing microspheres (MCGs) in the duodenal segment was superior compared to tablets and chitosan granules (5, 19).
In the present work, we report a pharmacokinetic study in adult dogs of the absorption of didanosine encapsulated in MCGs, which were recovered with the gastro-resistant polymer Kollicoat® MAE 100P. The study focused on a comparison among didanosine absorption from our MCGs, conventional didanosine tablets and commercial gastro-resistant chitosan granules (CG).
Materials and Methods
Materials. Didanosine-buffered tablets were provided by Farmanguinhos (Rio de Janeiro, Brazil); commercial gastro-resistant granules, Videx® EC, were donated as a gift from Single Health System (Campinas, Brazil); didanosine was provided by Labogen Química Fina e Biotecnologia S.A. (Indaiatuba, Brazil); Kollicoat® MAE 100P was donated as a gift from BASF (São Paulo, Brazil); acetic acid and sodium TPP were purchased from Synth (São Paulo, Brazil). Chitosan (approx. MW 296.6 kDa and deacetylation 82.83±3.63%) was obtained from Polymar (Fortaleza, Brazil). MicrocrystaIline cellulose was obtained from Henrifarma (Campinas/SP, Brazil). Distilled water was purified by a Milli-Q system (Millipore®, São Paulo/SP, Brazil).
Preparation and characterization of chitosan microspheres. The chitosan microspheres were prepared by ionotropic gelation of chitosan and sodium TPP as cross-linking agent, according to Santana et al. 2008 (17). Briefly, the microspheres were formed by dropping an aqueous solution composed of TPP (2 g/l), magnesium hydroxide (6 g/l) and didanosine (39 g/l) to an aqueous solution of chitosan (2.0% w/v) previously acidified with acetic acid (pH=5.5). The solutions were mixed under stirring (2,000 rpm) at 25°C.
The microspheres were characterized by the mean diameter and distribution through dynamic light scattering technique using a Malvern Autosizer 4700, (United Kingdom, UK, Malvern), and by the zeta potential using a Malvern Zetasizer 3000HSA (United Kingdom, UK, Malvern). The morphology and the surface of the freeze-dried microspheres were analyzed by scanning electron microscopy (LEO, LEO 440i model, ElectronMicroscopy/Oxford (Cambridge, England). The efficiency of the microspheres for incorporation of didanosine was calculated as the ratio of the difference between the initial amount of drug used in the preparation and the free drug present in the supernatant aqueous phase after centrifugation, to the total mass of drug. The results are shown as percentages.
Production, coating and characterization of the pellets. The pellets were prepared by extrusion and spheronization, according to Severino et al. (20). Briefly, the previously prepared microspheres were dried in an oven at 40°C until 75% humidity. The same chitosan 4.8% (by mass) and microcrystalline cellulose 4.8% (by mass) were added as excipient to the wet mass of the microspheres for the production of the pellets. The formulation was then extruded at 20 rpm using a Caleva extruder (20 model, Sturminster Newton, England). The extrudates were spheronized at 100 rpm for 5 min in a Caleva Spheronizer 120. The pellets were dried in a fluidized bed (Würster, Uniglatt, Glatt, Binzen, Germany) at 50°C inlet air temperature.
The pellets were also initially coated using Kollidon® VA 64 polymer using an aqueous solution 10% (m/v), and dried for 30 min to prevent aggregation, due to the porous surface of the granules. Kollidon® also facilitated the subsequent coverage, forming an impermeable surface by sticking a thin film of the gastro-resistant polymer. A second coating, the gastro-resistant coating, was employed as an alcoholic solution containing 30% (w/v) with Kollicot 100® MAE P, 1.5% (w/v) propylenoglycol, 0.5% (w/v) magnesium trisilicate and 0.5% (w/v) titanic dioxide. Both processes were performed in a Würster-type fluidized bed operating at an air flow rate of 6 m3/h, under inlet air temperature of 50°C and 0.9 bar. The dispersion was fed at 0.5 g/min through a peristaltic pump (Watson Marlow, Barueri, São Paulo, Brazil)
The pellets were characterized by size parameters through Feret diameter, sphericity and elongation measurements. Morphology was visualized by photograph obtained from a stereoscope (Motic, SMZ-161 model, British Columbia, Canada).
Animal protocols. The protocol used for didanosine pharmacokinetic evaluation was previously approved by the Ethics in Research Committee of University of Rio Grande do Sul (#2007760). A previous pilot study was carried out to demonstrate the applicability of the designed experimental protocol. Subsequently, the complete study was performed in male adult dogs (10 kg) which were previously vermifuged a week before the experiment and maintained individually. The dogs were fasted for 12 h before drug administration and received water and food 2 h later. Didanosine was evaluated using the three formulations: MCGs, tablets and the gastro-resistant Videx® EC. Each formulation was administered orally as a single dose of 500 mg didanosine to nine dogs. Blood samples (2 ml) were collected from the cephalic vase of the dog foot in heparinized VACUETTE® (Campinas/SP, Brazil) tubes before (time 0) and at 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, 10, 12, 18, 24 e 36 h after administration. The blood cells were removed by centrifugation for 10 min at 91 RCFxforce and the separated plasma was stored at −20°C until assayed.
The pharmacokinetic parameters were determined by the non-compartmental approach. The elimination rate constant (kel) was calculated by the log-linear regression of didanosine concentration during the elimination phase and the half-life (t1/2) was calculated as 0.693/kel. The maximum concentration of didanosine in the plasma (Cmax) and the corresponding time (tmax) were obtained by visual inspection of the concentration–time profiles. The area under the plasma concentration versus time curve (AUC0–t) from time zero to the time of last measured concentration (Clast) was calculated by the log-linear trapezoidal rule. The AUC zero to infinity (AUC0–∞) was obtained by the addition of AUC0–t and the extrapolated area determined by Clast/kel. The adsorbed fraction of drug in the bloodstream, considered the same bioavailable fraction (Frel) was calculated as AUC/(administered dose) and the corresponding medium oral clearance was CL/Fel, where CL is the clearance rate. The extrapolated area under the concentration–time curve (AUMC) was the volume taken from each of n samples to form a pooled sample, which is proportional to tn(tn+1 − tn−1), except at t0 where the aliquot volume is 0 and at tlast (the last time of collected sample). AUMC was calculated by Cpooled × T2/2, where T is the overall experimental time (tlast − t0). The ratio between AUMC and AUC yields the mean residence time.
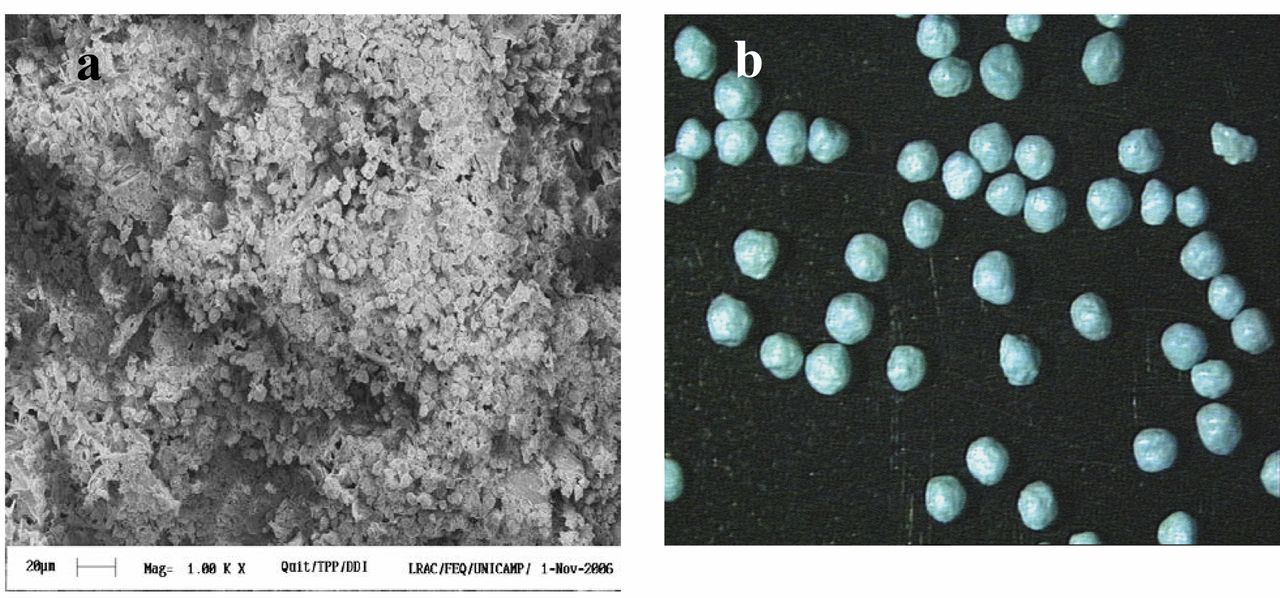
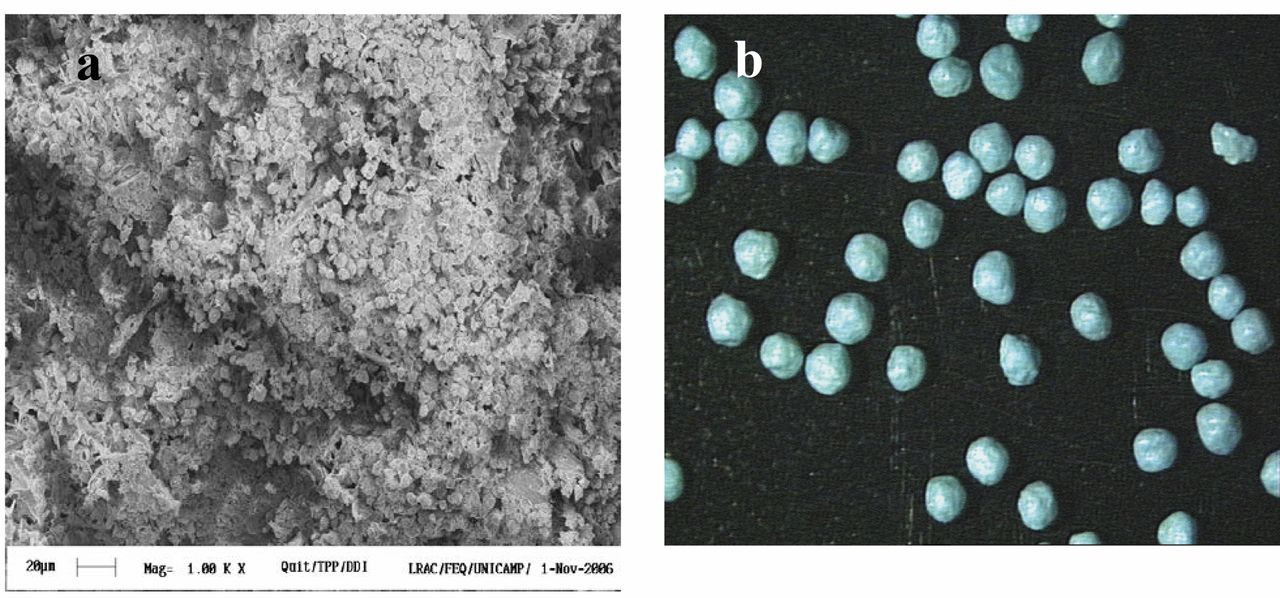
Morphologies of the chitosan microspheres encapsulating ddI (a) and the chitosan granules covered with the gastro-resistant polymer Kollidon® VA 64(b).
Analytical procedures in plasma. The protocol used for the analysis of didanosine in plasma was based on the analytical methodology described by Severino et al. (21). Previously, the samples were submitted to an extraction in a solid phase mounted in the Prospekt 2™ equipment (Spark Holland, Emmen-SPE, the Netherlands). The online SPE system was composed by the following modules: automated cartridge exchange (ACE) module for disposable cartridge exchange, high-pressure dispenser (HPD) for handling of solvents and an auto sampler Triathlon. The SPE cartridges were specified as BondElut C18, 40-90 μm of size particles and 10 mm×2 mm i.d. The samples were analyzed by high-performance liquid chromatography using a complete Shimadzu system (Shimadzu, Italia, Milan, Italy). The analytical column C18 (15 cm ×4 mm V04-745), was preceded by a guard column ACE® 5C18, V03-417, Analítica®. All samples and standard solutions were eluted at 28°C in a 1.0 ml/min flow rate, using a mixture of 0.02 M potassium phosphate buffer (KH2PO4), acetonitrile (96:4, v/v) and sulfonic acid heptane 0.5% (w/v) as the mobile phase, at pH 6.5 adjusted with triethylamine. Didanosine in the injected samples (50 μl) was detected at 250 nm wavelength.
Results
Physicochemical properties of the microspheres and granules. The mean diameter of the microspheres was 1±0.2 μm and the zeta potential was 64 mV. The efficiency of didanosine incorporation was 48.9%. The uncoated spheronized granules and the Kollidon® VA 64-coated ones were sized 0.90±0.15 mm and 1.05±0.26 mm, respectively. These are diameters by the industrial pharmaceutical technology recommended for filling conventional gelatin solid capsules used for administration of drugs. Figure 1 shows the morphologies of the microspheres and the coated granules.
Figure 2 shows the pharmacokinetic plasma profile after administration of didanosine-buffered tablets, CG and MCG granules. Table I summarizes the mean parameters obtained by the individual non-compartmental analysis.
Discussion
Didanosine-loaded microespheres were prepared by the ionic crosslinking interaction with the use of tripolyphosphate and chitosan. The crosslinking interaction is based on electrostatic interactions by chitosan protonated in amine group with TPP negatively charged anionic group (16). Microespheres are employed to improve the oral bioavailability of drugs (26), by enhancing the mucoadhesiveness in mucous tissue (3) and by active protection against degradation, obtaining more effective and less toxic formulations (23). The controlled release enables maintaining the blood levels drug for a longer period of time. Thus, the drugs remain above the level at which it is effective and below the level at which it is toxic. Chitosan was selected because it is a biocompatible and biodegradable polymer (7).
The microspheres studied in this work showed diameter of approximately 1 μm, zeta potential of 64 mV and EE of 48.9%. The zeta potential reflects the surface electrical charge of the particles, which is influenced by changes at the interface with the dispersing medium, due to the dissociation of functional groups on the particle surface or adsorption of ionic species present in the aqueous dispersion medium (18). The physical and chemical stability of polymeric systems emphasizes that in module, a relatively high value of the zeta potential is important for good stability, because large repulsive forces tend to avoid aggregation due to the occasional collisions of nanoparticles. Therefore, positive zeta potential increases the affinity towards the mucosa, characterized by the interaction positive electrostatic charges between protonated amine groups of chitosan with the negatively charged sialic acid present in mucin (5).
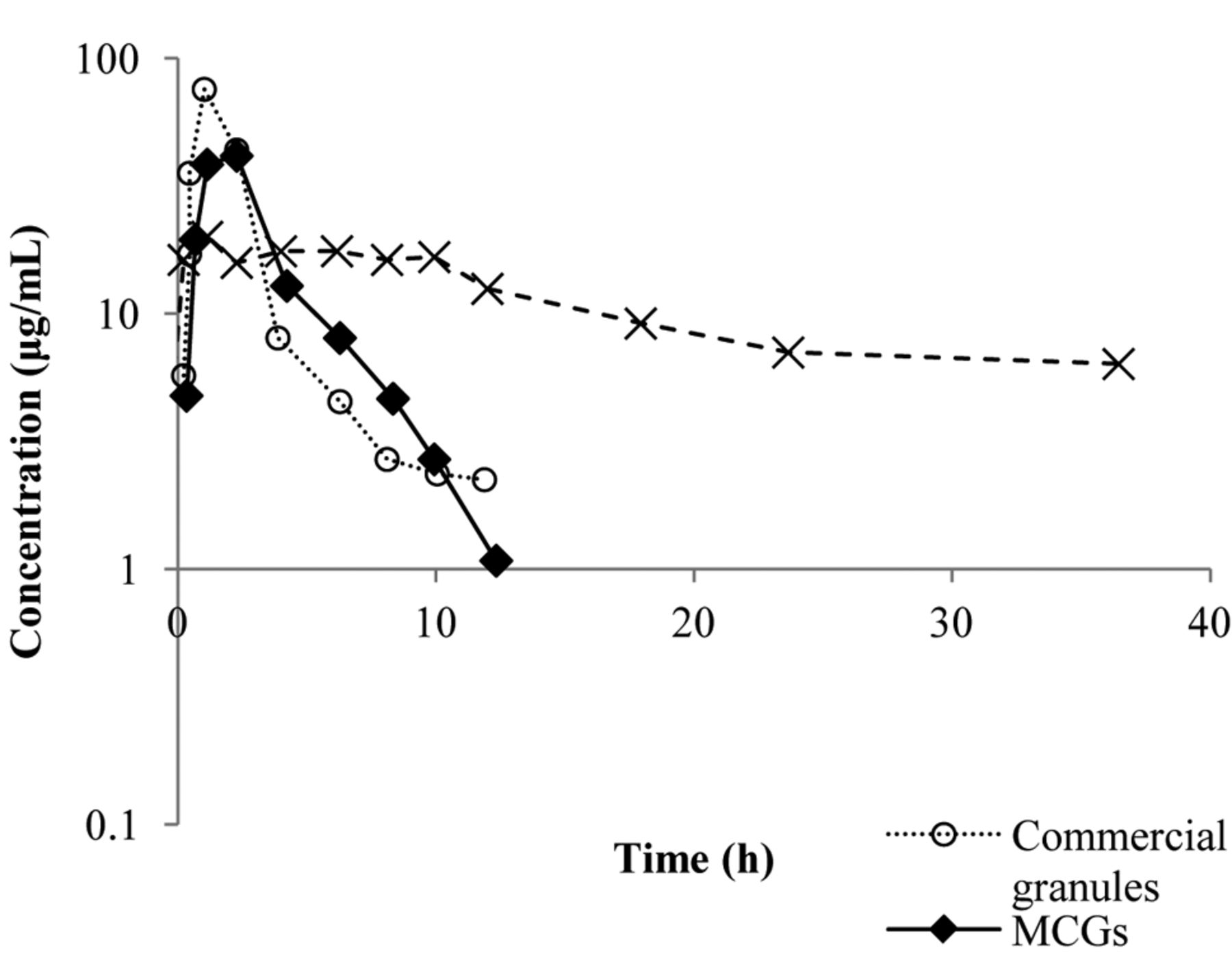
Pharmacokinetic profiles in the plasma of dogs of ddI from the different formulations: (○) commercial granules, (▴) buffered tablets and (*) MCGs.
The encapsulation efficiency (EE) is affected by many factors e.g. as drug characteristics, polymer concentration, drug polymer ratio, production process. The lower EE was influenced by high didanosine solubility in water (10), and low concentration of chitosan (22).
The morphology of microspheres is shown in Figure 1A. The microspheres are spherical in shape and had a rough surface. Ko et al., 2002 (8) produced chitosan microparticles and observed that the morphology is dependent on chitosan and TPP concentration, and pH. They obtained more spherical microspheres using at low pH values (pH 2.5) to prepare microparticles with TPP solution.
Figure 1B shows a spherical morphology of pellets with Feret diameter of approximately ~0.90 mm. The Feret diamenter is expected to be 0.9-1.1 mm because the diameter of the mold. In addition, the diameter is influencing by coating, type and quantity of excipients (chitosan and microcrystalline cellulose) added in the wet mass of the microspheres for the production of the pellets. Similar results were obtained by Steckel and Mindeermann-Nogly, 2004 (24), who produced chitosan pellets by extrusion/spheronization technology.
The results show the different plasma profile of our MCGs compared to the other formulations analyzed. Our granules do not show any peak concentration, as was observed for the buffered tablet and the commercial granules. Nevertheless, our granules showed a sustained delivery of didanosine over 36 h (Figure 2).
Pharmacokinetic parameters calculated through the non-compartmental analysis in the plasma of adult dogs. (Confidence level 95%)
The statistical analysis of the parameters t½, AUC0-∞, Cmax and tmax also shows a significant difference (with 95% confidence) in MCGs from the other evaluated formulations (Table 1). The AUC0-∞ of our MCGs was higher (59±11 μg·h/ml) than that for the tablets (20±5 μg·h/ml) and the CG (16±5 μg·h/ml). The half-life of our MCGs was 22.1±7.5 h, which is higher than that of the buffered tablets (5.4±2.4 h) and the CG (2.9±0.7 h). These values confirm the sustained delivery and absorption of didanosine from our MCGs, compared to the other formulations which were immediately delivered as shown in Figure 2. The Cmax and tmax. values were 9.1±3.0 μg/ml and 53.4±10.8 min; 4.5±1.2 μg/ml and 93.6±31.8 min for the tablets and CG, respectively. Instead of Cmax and tmax, our MCGs showed a plateau for the plasmatic concentration at 2.21±0.47 μg/ml which was extended for 36 h after the oral administration. Figure 1 also shows that the plasma concentration of didanosine released from the tablets and CG was drastically reduced in 12 h.
The amounts of absorbed didanosine were similar for 12 h for all the evaluated formulations. Considering the tablets as a bioavailability reference (F=1), the relative bioavailability (Frel) in 12 h of CG and MCGs were 1.11 and 0.85, respectively. In contrast, the bioavailability of didanosine from our MCGs was prolonged for 36 h and the amount absorbed was approximately twice that of the tablets (Frel=2.17).
The extrapolation of AUC (Table I) identifies the time for elimination of the total drug in the study. Values lower than 20% are considered adequate in the literature for the oral administration of drugs in pharmaceutical forms of immediate delivery, as shown for the tablets and CG. The sustained delivery of didanosine from our MCGs was also confirmed by an AUC value higher than 20% (33±10%). Figure 2 shows that the elimination phase of the drug is just starting at 36 h leading to a significant change in the half-life after use of granules. The granules were changing the process elimination of the drug rather than the absorption process, the clearance should be changed. Table I depicts the relative values of the clearance absolute bioavailability (CL/F). If assuming that intravenous drug administration has the bioavailability of conventional form, it is equal to 1 and the relative bioavailability (Frel) of the other formulation refers to their availability absolute values as 1 of Frel. This proves that the granules are only interfering with the process of absorption of ddI.
From the plasma profiles and pharmacokinetic parameters assessed in this study, it appears that we have succeeded in producing a controlled release formulation. Thus, the developed formulation will be advantageous to patients by probably increasing the interval between doses, facilitate administration to children and people with limited swallowing and providing better patient compliance. Furthermore, the proposed formulation does not cause the related side-effects of magnesium and aluminum hydroxide observed with conventional formulations.
Sustained release occurs by adhesion of chitosan intestinal membrane through the connections between the positively charged chitosan and the negative sialic, acid present in the intestine, which provides better absorption by increasing the permeation.
Conclusion
The plasmatic profiles and the calculated pharmacokinetic parameters point to the MCGs as a promising formulation for in vivo improvement of absorption of didanosine. These improvements are due to the sustained release and mucoadhesiveness of the MCGs. Furthermore, the MCGs benefits didanosine administration to children and elderly people because it allows fractionation of the dose and ease of swallowing. The MCGs also benefit taste because the granules are coated and their size allows their administration inside gelatin capsules. The buffered tablets are chewed, leaving a metallic taste in mouth. Finally, the MCGs contain 10% buffer in the formulation, reducing or avoiding the collateral effects imparted by the buffer in the conventional tablets (about 50%). All of these advantages promote the compliance of patients to their therapy due to convenience and comfort. Moreover, MCGs are cheap, due to the low cost and abundance of the raw materials and their production is easy, reproducible and scalable.
Acknowledgements
The Authors acknowledge the financial support from the Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP/Brazil, #20801-2), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes) and the Conselho Nacional de Pesquisa (CNPq, Brazil). The Authors also wish to acknowledge Fundação para a Ciência e Tecnologia do Ministério da Ciência e Tecnologia, under reference ERA-Eula/0002/2009.
- Received July 27, 2014.
- Revision received September 13, 2014.
- Accepted September 16, 2014.
- Copyright © 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.