


Abstract
Recent immune data on vitamin D3 deficiency help to more clearly understand chronic fatiguing illnesses, such as autoimmune disorders, cancer and chronic fatigue syndrome (CFS). The vitamin D3 pathway is activated by stress and requires sufficient stores of precursor 25-hydroxyvitamin D3 for proper cell and immune functions. In vitamin D3 deficiency, secretion of the antimicrobial peptide cathelicidin is reduced, leading to impaired auto/xenophagy. As a result, phagocytosis, cytotoxicity, antigen processing and antigen presentation become dysregulated. In addition, vitamin D3 deficiency affects T- and B-lymphocyte activation, as well as quantity, maturation and function of regulatory natural killer T-cells and their counterparts in the gut, i.e. T-cell receptor-αβ, cluster of differentiation-8αα-positive intraepithelial lymphocytes. Consequently, innate and adaptive immunity become de-regulated, with microbial effects contributing further to this. Persistent infections, chronic inflammation and fatigue follow. Vitamin D3 substitution in such conditions may help to prevent or to ameliorate such chronic conditions, even in patients with cancer.
Vitamin D3 and calcium deficiency are found in various diseases, including immune disorders (1-6) and in conditions with chronic fatigue (7-15). Some positive vitamin D treatment reports (11, 12, 15, 16), indicate a possible connection between vitamin D3 deficiency and chronic fatigue, exhaustion and depression. This article reviews immune reactivity as related to vitamin D3 levels, and energy de-regulation in vitamin D3 deficiency.
Sufficient Supply of Vitamin D3 is Important for Proper Human Cell Functions and Stress Response
Following light activation in the skin and further enzymatic processing, the active metabolite 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] of vitamin D3-precursor, also called calcitriol, is synthetized from the immediate pro-hormone 25-hydroxycholecalciferol (25OHD3) by the enzyme cytochrome p450-hydroxylase27B1 (CYP27B1). The reaction is mediated in the kidneys by parathormone and is calcium-dependent (17). This endocrine pathway serves in the tight regulation of serum calcium levels (5, 6, 17). However, most cells also convert 25OHD3 to active 1,25(OH)2D3, which serves as a para-, or autocrine transcription factor binding to many gene loci (17-22). In addition, 1,25(OH)2D3, in an epigenetic way, directly influences cell signals and cell functions (21, 23-25). 1,25(OH)2D3 is an important cell regulator and influences cell development, differentiation, proliferation and cell-cycle control (20, 21). Various kinds of cell stress cause activation of the vitamin D pathway, and generation of 1,25(OH)2D3 requires sufficient supply of the precursor 25OHD3 in order to establish an effective protective response (17, 22).
In particular, immune functions are highly dependent on 1,25(OH)2D3. Adequate functioning of immune cells depends on the vitamin D3 pathway as initiated by the expression of vitamin D receptor (VDR) and vitamin D-activating enzyme CYP27B1 (1, 5, 17, 26-29). In addition, 1,25(OH)2D3 mediates the induction of voltage-gated chloride and calcium ion channels, regulating the secretion of cellular products, e.g. transmitters and immune granules (23). The complex interplay of vitamin D3-induced effects leading to immune effectiveness and to balanced immune reactions are summarized in Figures 1, 2 and 3.
Physical and Functional Epithelial Barriers are Enhanced by 1,25(OH)2D3
At the first stage of defense at dermal and mucosal barriers, 1,25(OH)2D3 regulates gene expression of major proteins responsible for sealing epithelial tight junctions, i.e. claudins, thus stabilizing skin and mucosal barriers (28, 30-32). 1,25(OH)2D3 enhances keratin differentiation (33-34), modulates mitogen-activated protein kinase-signaling in keratinocytes by exerting anti-inflammatory and protective effects (35), and down-regulates matrix metalloproteinase-9 (36). Also 1,25(OH)2D3 protects against radiation effects (37), and against programmed cell death in stressed keratinocytes (38). 1,25(OH)2D3 reduces the responsiveness of interleukin-2(IL-2)-activated T-lymphocytes, thus diminishing stress-induced local inflammatory reactions (39).
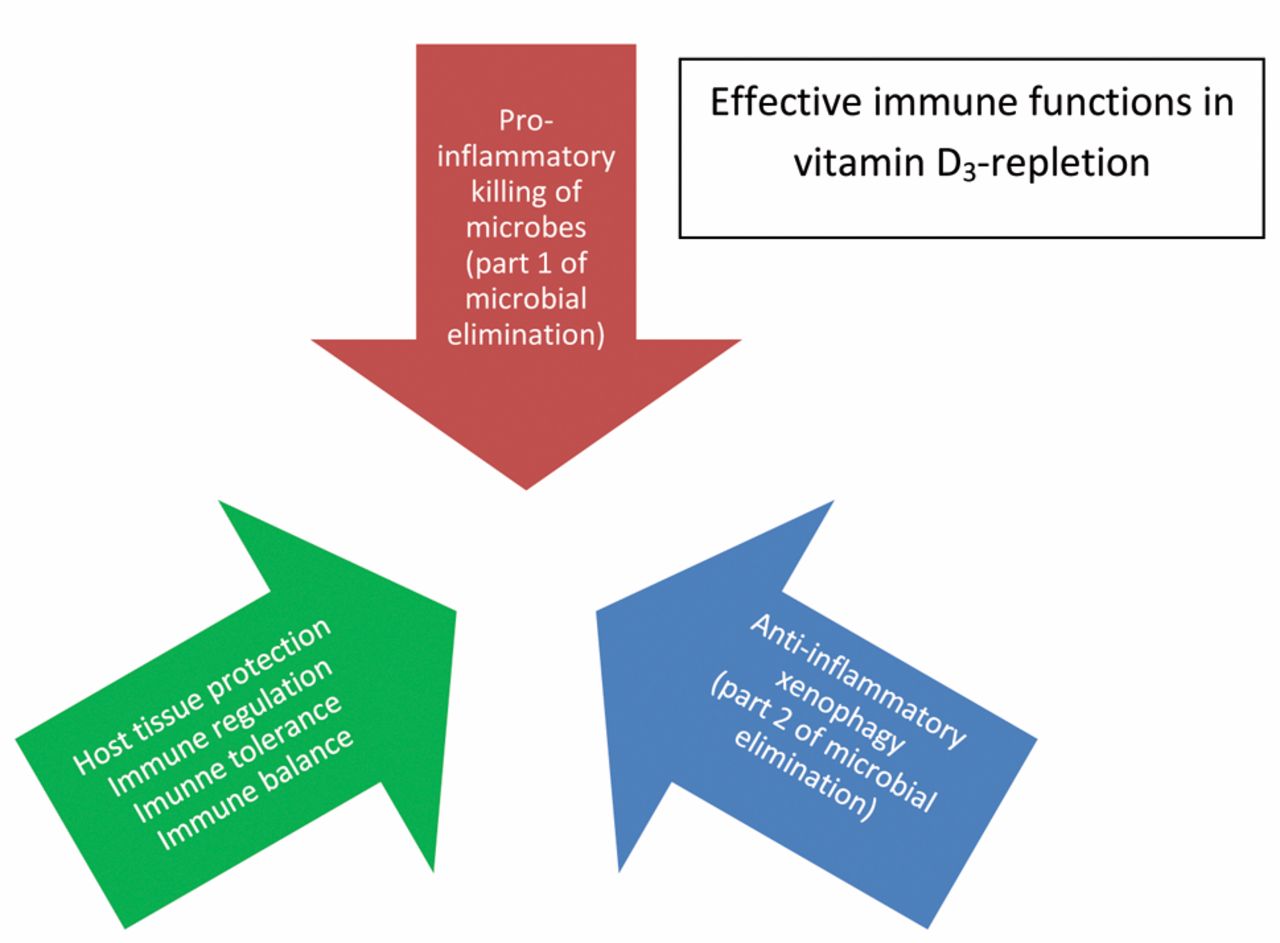
Vitamin D3 repletion ensures effective microbial elimination, yet combined with prompt protective anti-inflammation and immunoregulation.
Influence of 1,25(OH)2D3 on Antimicrobial Peptides (AMiPs)
AMiPs are produced after a microbial challenge by epithelial cells, by natural killer cells (NK), γδ-T-lymphocytes, and also by B-lymphocytes (28, 29, 40, 41) serving as important biochemical barriers. AMiPs de-stabilize bacterial membranes by cationic and electrostatic effects (28, 29, 40). In addition, they are multifunctional and bind to certain cell signaling receptors and to DNA (42-45). They interact with immune, endothelial and epithelial cells (42, 46, 47), enhancing phagocytosis, auto/xenophagy, cellular cytotoxicity, chemoattraction of immune cells, induction of memory T-cells, angiogenesis and wound healing (28, 29, 40-42, 46, 47). AMiPs also possess immunoregulatory effects by suppressing pro-inflammatory cytokines, down-regulating toll-like receptor (TLR) expression, and by neutralizing endotoxins (27, 28, 42, 46, 47).
In humans, 1,25(OH)2D3 significantly enhances the expression of two antimicrobial peptides, called cathelicidin and defensin-4B (27-29, 40, 49, 50). Importantly, AMiPs act synergistically with 1,25(OH)2D3, enhancing innate immune functions, as well as down-regulating inflammation, and adaptive immune regulation. In a counter-measure, bacterial toxins reduce cathelicidin expression (51). Interestingly, in contrast to humans, mice devoid of sun exposure, regulate cathelicidin expression independently of 1,25(OH)2D3 (52).
Secretory immunoglobulin A (sIgA), another biochemical barrier of skin and mucosa, is supported by 1,25(OH)2D3 rather indirectly by inducing the expression of immunoglobulin A fragment crystallizable (IgA Fc) receptor on phagocytes leading to enhanced binding of sIgA (53-55). Furthermore, 1,25(OH)2D3 induces the C-C motif chemokine receptor-10 (CCR10) in human B-cells, resulting in enhanced B-cell differentiation to IgA-secretory cells, with potential for homing to the gut (56, 57).
Auto/Xenophagy as an Important Cellular Rheostat and its Relation to 1,25(OH)2D3 and Cathelicidin
Autophagy, in infection also named immuno- or xenophagy, is essential for cell and immune functioning (58-62). Damaged material (e.g. cell or tissue) is degraded in a multistep process, with its products being used for functional adaptation, recycling of building blocks, and energy production (58, 60). Autophagy functions like a rheostat, linking internal and external conditions to cell-regulatory pathways (60, 63).
Distinct autophagic steps (initiation/induction with nucleation; elongation and closure of the autophagosomal double-membrane; maturation and fusion with the lysosome), generate the auto(phago)lysosome, which degrades or extrudes the ingested material with greater efficacy than the phagolysosome (52, 58, 60, 62, 64). Three cell signaling systems initiate auto/xenophagy. Firstly, inhibition of mammalian target of rapamycin (mTOR)-AuTophaGy-related-1 (ATG1) complex; secondly, the beclin1/class III phosphoinositol-3 kinase C3/vacuolar protein sorting associated protein (PI3KC3)/VPS34) complex (58, 65); and thirdly, the AuTophaGy-related proteins ATG5-ATG12-ATG16L1 and ATG7-ATG3-ATG8/microtubule-associated protein 1A/1B-light chain 3 (LC3)/gamma-aminobutyric acid receptor-associated protein complex (GABARAP) (59). These signal systems respond to stress by activating TLRs, or nucleotide-binding oligomerization domain (NOD)-like receptors, nuclear factor-κB (NF-kB), and pro-inflammatory cytokines, such as interferon-gamma (IFN-γ) and tumor-necrosis factor-alpha (TNF-α), as well as by elevating intra- or subcellular calcium with subsequent activation of adenosine monophosphate-activated protein kinase (AMPK) (59, 63, 66, 67).
Importantly in autophagolysosomes, self- and non-self-peptides are joined to antigen-presenting molecules (58, 62), a process contributing substantially to immune control, anti-inflammatory activity, immune memory (59, 62, 68-70), and induction of self-tolerant T-cells (58, 65). Thymic epithelial cells and thymocytes use effective auto/xenophagy for positive and negative selection of T- and B-cells (71). De-regulated autophagy has been reported to result in autoimmune disease (62, 64) and cancer (63, 72).

The most important direct and indirect cooperative, and in part, bi-directional immune effects of vitamin D3 pathway. 25-OHD3 25-Hydoxyvitamin D3; 1,25(OH)2D3 1α,25-Dihydroxyvitamin D3; VDR vitamin D receptor; mDC myeloid dendritic cell, NK T-cells Natural Killer T-cells.
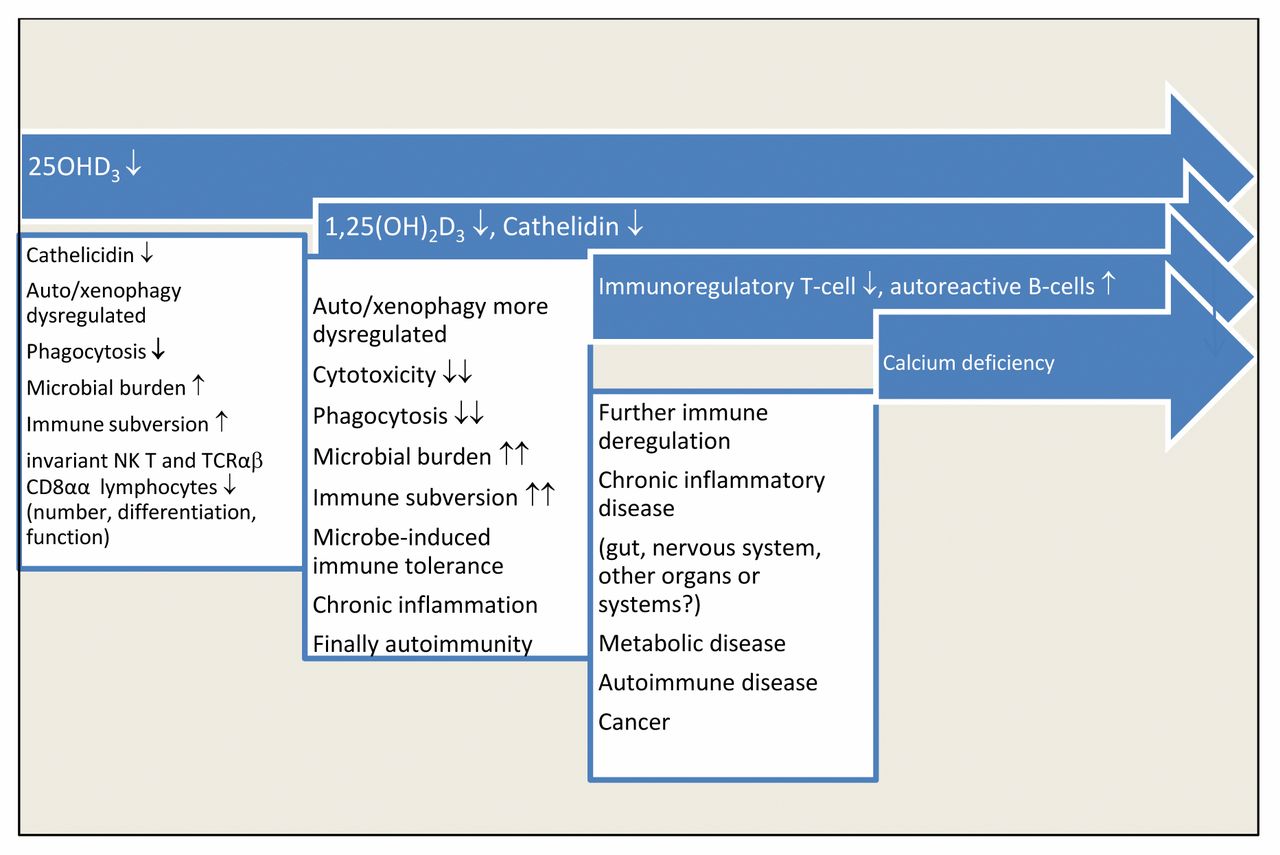
Vitamin D3-depletion results in a substantial innate immune defect, whereas the expected adaptive immune intolerance presumably is modified by microbial immune subversion mechanisms, characterized by marked immune tolerance, in spite of smoldering chronic inflammation. A hierarchy of further immune deregulation steps will ensue, culminating in the problem of chronic calcium deficiency with exacerbation of immunoderegulation.
1,25(OH)2D3 enhances auto/xenophagy at multiple levels, such as NOD2 receptor expression, ATG16L recruitment to the site of bacterial entry (73), or PI3KC3 activation, thus supporting initiation, nucleation and elongation of the autophagosomal membrane (64).
However, what is more efficient in auto/xenophagy, is the cooperation of both 1,25(OH)2D3 and cathelicidin (26, 49, 52, 64, 66, 67, 74, 75). Both agents enhance beclin-1 gene expression (66, 75, 76), and promote autophagosome maturation, as well as lysosomal fusion (26, 64, 74). They also increase acidity and protease activity in autophagolysosomes (66, 74). Interestingly, deranged autophagy compromises cathelicidin expression, revealing bi-directional coupling between cathelicidin and auto-xenophagy (64).
Auto/xenophagy further depends upon negative control mechanisms (77). Negative control regulators include members of the autophagy machinery itself such as ATG16L1 and ATG5-ATG12 complex, resulting in reduced production IL-1β, IL-18 and type I IFN-α (58, 62, 77). 1,25(OH)2D3 contributes to the negative control by inhibiting pro-inflammatory signals such as NF-kB, TNF-α, IFN-γ, and by promoting the cyclin-dependent kinase inhibitor P19INK4D (64).
Endo- and Phagocytosis are Enhanced by 1,25(OH)2D3 and Cathelicidin
1,25(OH)2D3 enhances endocytosis by augmented gene expression of antigen uptake receptors, including mannose receptor and FC-γ receptor II (CD32) (74, 78). Phagocytosis is promoted by 1,25(OH)2D3-mediated enhancement of macrophage maturation, lysosomal production of acid phosphatases and hydrogen superoxide (H2O2), and by enhanced xenophagy (1, 5, 28, 50, 79).
Although promoting endocytosis and phagocytosis, 1,25(OH)2D3 reduces antigen presentation, T-cell activation, and secretion of pro-inflammatory cytokines (80, 81). Cooperation of 1,25(OH)2D3, cathelicidin and auto-xenophagy optimizes neutralization of the endotoxin-induced pro-inflammatory TNF-α and nitric oxide production (42, 52, 75, 82).
1,25(OH)2D3 Induces Immune-tolerant Myeloid Dendritic Cells and Regulatory T-Cells
Innate and adaptive immunity are linked to dendritic cells (DCs) that orchestrate adaptive immune responses and anti-inflammatory regulation (56, 83, 84). The best known DC subtypes are myeloid (mDCs) and plasmacytoid (pDCs) dendritic cells (56, 83, 85).
mDCs are antigen-presenting cells (86) and carry antigen-binding surface receptors of the major histocompatibility (MHC) group (83, 86, 87), as well as MHCI-like-receptors, such as the glycoprotein cluster of differentiation protein-1 family member (CD1d) (83). Activated mDCs secrete IL-12 (87, 88), and induce distinct T-cell populations such as Th1-cells for intracellular and Th17-cells for extracellular pathogens, and Th2-populations with regulatory T-cells (Treg) for incompletely-destroyed pathogens (86, 87).
Without activation, mDCs are highly dependent on 1,25(OH)2D3, which induces substantial immune tolerance (27, 56, 83, 84, 87, 89, 90). Immature mDCS, not yet stimulated, and their precursor cells express abundant VDRs, in contrast to stimulated mDCs. Hence, more differentiated mDCs, with less VDR expression, preserve their required defensive potential (56, 84, 88). However, even on antigenic stimulation, mDCs are more tolerant when 1,25(OH)2D3 is present (56, 84, 88, 89).
1,25(OH)2D3-induced immune tolerance is brought about by down-regulation of pro-inflammatory molecules in mDCs, such as CD40, CD80, CD86, MHCII, CD54, IL-12/IL-23p40 and the C-C motif chemokine ligand 17 (CCL17) (56, 84, 88). In addition, immune-inhibitory molecules are up-regulated including immunoglobulin-like transcript-3 (ILT3) and programmed death ligand-1 (PDL-1) (56, 84). As a result, mDCs secrete less IL-12, but more IL-10 and transforming growth factor (TGF) (26, 56, 91), thus promoting a shift from Th17 and Th9 to regulatory CD4+CD25+ Tregs (3, 92, 93). Tregs express increased inhibitory receptors including FOXP3 and cytotoxic lymphocytic antigen-4 (CTLA-4) (27, 84, 94, 95). Of interest is the mutual tolerance induction observed between DCs and Tregs (56, 96). However, the induction of a significant Th2-polarity by 1,25(OH)2D3 has been recently challenged (97).
1,25(OH)2D3-induced T-cell tolerance is also mediated by direct action on activated T-cells (26, 56, 98, 99). 1,25(OH)2D3 reduces the proliferative activity of Th1 cells by reducing the expression of pro-inflammatory cytokines such as IL-2, IFN-γ and TNF-α (56, 98-100). 1,25(OH)2D3 also enhances T-cell secretion of IL-4 and IL-13 (90) by inducing the stress and translation inhibitor protein cytidine-cytidine-adenosine-adenosine-thymidine box motif(CCAAT)/enhancer-binding protein (EBP) homologous protein (CHOP) (101, 102), and induces important Th2 transcription factors, such as signal transducer and activator of transcription-6, and GATA-binding protein, and CD 200, an immunoglobulin-like molecule on CD4+ T-lymphocytes, which inhibits Th17 differentiation (103).
Protective mucosal tolerance is also induced by 1,25(OH)2D3 by enhancing mucosal homing of immunoregulatory dendritic cells (1, 28, 104-106). Stress- and 1,25(OH)2D3-induced induction of vitamin D-up-regulating protein (VDUP1) additionally suppresses pro-inflammatory lymphocytes in the lamina propria (107).
In contrast to mDCs, pDCs are specialized for virus control via IFN-α secretion (56, 84, 89). They also induce Tregs, yet appear to be independent of 1,25(OH)2D3 (56). pDCs induce Tregs and adaptive immune tolerance in several ways: i) by up-regulation of inducible co-stimulatory ligand (ICOSL) with or without secretion of indoleamine 2,3-dioxygenase (IDO) (84, 108), ii) by up-regulated expression of IL-27, IL-10, and TGF-β1-mediated inhibition of Th17 polarization (84, 85), iii) by secretion of vasoactive intestinal polypeptide (VIP) in conditions such as chronic inflammation and/or autoimmunity (109), and iv) by thymic stromal lymphopoetin (TSLP) by pDCs or epithelial cells (84, 110). Tregs are also induced by retinoic acid (84, 111).
Cathelicidin and NK cells in Innate and Adaptive Immunity
NK cells are activated by direct DC/NK contact and cooperate with CD8+ cytotoxic T-cells (112, 113). Further activating signals in a bi-directional way may be received from and sent to macrophages, polymorphonuclear leucocytes, T- and B-lymphocytes, mast and epithelial cells, or as inhibitory signals from Tregs (112, 114, 115, 116). NK cells secrete antimicrobial peptides such as α-defensin and cathelicidin (48, 115, 117) and induce anti-inflammatory, immunoregulatory (118) and immune-memory effects (112, 113, 115). Their maturation and function depend upon the environmental signaling milieu (112-114, 118, 119). However, reliable in vivo evaluation of NK cell functions is limited by complex cell kinetics and variable signals from the surrounding environment (114, 120, 121). ‘Exhausted’ NK effector cells were described in chronic infectious processes (113).
Due to these complex relationships, reported effects of 1,25(OH)2D3 on NK cells remain contradictory. Some reports describe inhibitory effects (122), with depressed NK cell activation and cytotoxicity in rats (123-125), reduced NK cell chemotaxis against eosinophils, and reduced IL-15-induced IL-8 secretion (126). Others showed inhibition of NK cell activation by 1,25(OH)2D3, but no inhibition of cytotoxicity, with reversed inhibition after immune activation with IL-2 secretion or exogenous IL-2 addition (127, 128).
In apparent contrast, enhanced NK cell cytotoxicity was observed following treatment with active vitamin D3 (calcitriol) in patients on hemodialysis (129). Enhanced NK cytotoxicity towards cancer cells resulted from 1,25(OH)2D3-induced increase of cathelicidin (46, 48). Indirect positive effects on NK cytotoxicity were mediated by 1,25(OH)2D3-induced increase of glutathione synthesis (130, 131), and by 1,25(OH)2D3-induced increase of extracellular calcium levels with elevated activity of protein kinase C (PKC) and N-alpha-benzyl-oxycarbonyl-L-lysine thiobenzyl ester (BLT) esterase (132). In addition, NK cell differentiation and maturation was augmented by VDUP1 expression, an effect that could be further enhanced by differential cellular calcium influx (107, 133).
Vitamin D Receptor and 1,25(OH)2D3 Are most Important for Invariant NK T-cells
NK T-cells express NK and T-cell type-specific receptors (134, 135). Most NK T-cells belong to the subgroup of invariant NK T-cells, also called class I NK cells (110, 136-139), that resemble functionally of Tregs rather than NK cells (134, 135). They are regarded as being most essential for overall immune balance (134, 140, 141). The term ‘invariant’ refers to their semi-invariant special T-cell receptor (TCR), with an invariant alpha and a restricted beta chain (140). Differently from conventional T-cells, they constitutionally secrete IL-4 and IFN-γ, and augment secretion rapidly after immune challenge (134-136).
IL-4 is important for immune B-cell activation (142) and prevents immune overstimulation, chronic inflammation and autoimmunity (136). In contrast, IFN-γ is important for viral clearance, further immune activation, antimicrobial defense (143, 144) and phagosome maturation (52).
Like NK cells, invariant NK T-cells exert both immune-activating and immunoregulatory activity, and link innate and adaptive immune functions by a mutual and multi-dimensional cross-talk (145-148). They augment cytotoxic CD8+ T-cell responses by induction of CD70 expression on dendritic cells (147). Invariant NK T-cells consist of several subgroups with functional differences (145, 146). Their numbers appear reduced in certain autoimmune diseases (145, 146). Invariant NK T-cells are tightly connected to the vitamin D pathway (100, 136-139). Development and function of a double-positive intra-thymic invariant NK T-cell precursor depends exclusively on intra-thymic VDR expression and VDR-dependent induction of the non-classical MHCI receptor CD1d (100, 136-139). CD1d is structurally associated with β2-microglobulin, similar to the MHCI receptor (134, 140). CD1d receptor presents self-antigens, preferentially endogenous lipids and glycolipids (136, 138, 141). Invariant NK T-cells are self-reactive, but not self-destructive (134, 136-138). Whereas agonist selection of invariant NK T-cells is completed in the thymus, full maturation is completed in the periphery where invariant NK T-cells preferentially inhabit the liver and spleen (140, 141, 143, 145).
During their specific differentiation steps, invariant NK T-cells lose CD8, often also CD4 co-receptor (100). They begin to express NK lineage receptors, such as the activating type II integral membrane protein receptor (NKG2D), members of the Ly-49 family, and finally the natural killer (NK) cell-associated marker NK1.1 (CD161) (138) and the T-cell memory CD44 receptor (137). During these maturation steps, immunoregulatory properties with protection against pathogens, cancer and autoimmunity are acquired (145, 146). Interestingly, not only 1,25(OH)2D3, but also VDUP1 is required for invariant NK T-cell development (133).
Studies on mice with a knocked-out VDR revealed reduced invariant NK T-cell numbers and reduced IFN-γ and Il-4 secretion after antigen challenge (100, 136, 149, 150). Cell maturation was compromised, with inability to up-regulate CD44+ and NK1.1+ receptors (100, 138, 149-151). Vitamin D3-deficient wild-type mice had reduced numbers of invariant NK T-cells due to increased apoptosis in the thymus, yet after vitamin D3 supplementation had almost normal cellular function (100, 106, 138, 150). However, substitution of 1,25(OH)2D3 did not fully-restore invariant NK T-cell numbers, an effect even transferred to their offspring, possibly due to epigenetic changes induced by vitamin D3 deficiency (150).
Both VDR- and vitamin D-deficient animals were prone to develop inflammatory bowel disease and experimentally induced encephalomyelitis (32, 100, 106, 149).
Intra-epithelial CD8αα TCRαβ Cells are Invariant NK T-cell-equivalent gut Mucosa Cells and Essential for Local Immune Balance
In the gut epithelium, a cell population has been found to functionally resemble invariant NK T-cells (32, 100, 106, 136). They also develop from the same intra-thymic invariant NK T-precursor cell. Like invariant NK T-cells, they depend upon intra-thymic agonist selection, they are self-reactive without self-destruction, and exhibit phenotypes of regulatory or memory cells (32, 100). In contrast to invariant NK T-cells, they express a gut-specific homodimeric CD8+αα chain in the presence of IL-15 (152), and are identified as TCRαβ+CD8αα+ intraepithelial lymphocytes (100, 106, 136).
Gut mucosa of VDR-knockout animals contains only half as many CD8+αα cells, and the CD4/CD8αα intraepithelial lymphocytes are totally absent, presumably due to failed gut homing (3, 100, 106, 136).
Activated B-cells Express VDR, and B-cell/invariant NK T-cell Interactions Modulate Immune Responses
Activated B-cells, like activated T-cells, express VDR. Mediated by their antigen-specific B-cell receptor, B-lymphocytes also present antigens and support phagocytosis (153, 154). Finally, activated B-cells secrete cathelicidin, and by interaction with 1,25(OH)2D3 and cathelicidin, they contribute to optimal immune defense and balance (155, 156).
1,25(OH)2D3 directly inhibits B cell proliferation by stabilizing the cyclin-dependent kinase inhibitor p27 (157). It also inhibits the differentiation of ‘post-switch’ memory B-cells and plasma cells, and reduces immunoglobulin production and secretion, e.g. by inhibition of CD40 signaling (56, 157, 158), particularly of IgE (158). 1,25(OH)2D3 promotes B-lymphocyte apoptosis, IL-10 secretion, and expression of CCR10 (56, 157).
Of importance, several types of B-cell/invariant NK T-cell interactions have been reported. Firstly, invariant NK T-cells support B-cell antibody production and proliferation of memory B-cells, even without CD4-T-cell help (159). Secondly, invariant NK T-cells reduce proliferation and promote apoptosis of splenic self-reactive, CD1d- and IL-18-expressing marginal zone B-cells (MZBs) possessing innate autoimmune potential (160-163). Thirdly, invariant NK T-cells enhance proliferation of immunoregulatory follicular B-cells (162), while conversely, MZBs and DCs activate invariant NK T-cells (164).
In addition, high surface-expression of CD1d on immature B-cells appears essential for the proliferation and differentiation of invariant NK T-cells (165). Patients with systemic lupus erythematodes (SLE) have a B-cell-specific subcellular transport defect of CD1d causing reduced amounts of surface CD1d. They also have reduced invariant NK T-cell numbers with diminished IL-2 stimulation and diminished IFN-γ and TNF-α secretion, while IL-10 secretion is augmented (165). This transport defect was not found in other immune cells. An intrinsic invariant NK T-cell defect was excluded in these experiments. For unknown reasons, normal CD1d surface expression on other cell types, such as cortical thymocytes, lymph node mantle zone and spleen MZBs, and resting monocytes could not compensate for this SLE-specific intrinsic B-cell defect (165). Most interestingly, patients with SLE who responded to rituximab treatment showed restored CD1d characteristics in immature CD1dhi B-cells and normalization of invariant NK T-cell numbers, activation and function (165).
Discussion
As shown here, vitamin D3 levels, metabolism and physiological immunoreactivity are intimately related. Insufficient levels and activities of D3 can cause immune dysregulation, resulting in various diseases, and can negatively influence the course of a variety of diseases.
Initial symptoms of low 25OHD3 levels are intermittent fatigue and recurrent infections which remit seasonally or after holiday. Insidiously, chronic fatigue syndrome may develop over time, typically promoted by stressful and exhaustive life conditions, infections, traumatic or toxic injuries. Hallmarks of chronic fatigue syndrome/myalgic encephalopathy (CFS/ME) are severe and disabling fatigue, absence of fever in spite of general malaise resembling an acute infection, and exertion-induced aggravation of functional disabilities, as well as many additional symptoms, in particular generalized pain, sleep disorder, and gastrointestinal discomfort. Obvious organ damage is lacking, whereas reactive depressive symptoms prevail. Symptom shift to fibromyalgia (FMS) seems to be the rule when patients get older. Typically, FMS is correlated with chronic skeletal disorders of low inflammatory activity and neuropathic pains. Yet many people do not acquire CFS/ME or FMS. They suffer from clear-cut diseases that are supposed to be triggered by vitamin D3 deficiency or insufficiency. Usually, disabling fatigue accompanies chronic inflammatory and autoimmune diseases, as well as cancer, whereas less severe fatigue is usually reported by patients with chronic tissue de-generation. Severe chronic fatigue has also been observed in psychiatric diseases. Often, patients view fatigue as the most disabling among all the other disease symptoms.
Although clues are emerging that low 25OHD3 may cause chronic fatigue, altered lifestyle and behavior would also explain it, in particular with respect to patients who appear depressive or exhausted. Additionally, measurement of chronic fatigue is highly subjective. However, the diagnosis of depression or exhaustion is also subjective, in particular when the differential diagnosis of CFS/FMS or FMS is not considered. In contrast, in chronic inflammatory, autoimmune or malignant diseases, physicians appreciate fatigue undoubtedly as being disease-induced. In order to overcome usual prejudice against chronic fatigue, it should be considered that inflammation may not only induce fatigue, but also alteration of mitochondrial function, auto/xenophagy, or excitation-metabolism coupling due to lowered subcellular calcium stores (22, 166, 167).
Fortunately, an increasing number of authors acknowledge the importance of vitamin D in immunoregulation (168-180). Epidemiological studies report a correlation between an insufficient level of 25OHD3 and several immune diseases, such as chronic pulmonary infections (168-170), multiple sclerosis (171-174), SLE (175, 180), diabetes and cardiovascular diseases (22, 176), and cancer (7, 177-179). Additionally, low 25OHD3 levels and elevated immunoreactivity against Epstein-Barr virus were found before the onset of multiple sclerosis (171), and up-regulation of VDR and CYP27B1 was found in active lesions (174). Low 25OHD3 levels also correlated with recurrence of spinal inflammatory lesions (172), and reduced survival in ovarian cancer (179). Genetic variations in enzymes of the vitamin D pathway were found to augment the risk for multiple sclerosis (173), and differentiated thyroid carcinoma (178). Low 25OHD3 levels were also prevalent in patients with FMS and CFS (2, 6, 8, 9, 11, 12, 14, 15).
In contrast to these epidemiological studies, interventional studies are still rare and small-sized. After one-year treatment with 2,000 IU (50 μg)/day cholecalciferol, inflammatory and hemostatic markers and disease activity in SLE improved (180). A dose of 800 IU (20 μg) cholecalciferol/day applied for 2.5-10 months improved fatigue in patients with myasthenia gravis (10). Rehabilitation outcomes improved after vitamin D supplementation in those with multiple illnesses (16). 4,000 IU (100 μcg) cholecalciferol/day for one year reduced recurrent infections of the respiratory tract significantly (169). Importantly, a pilot study showed clearly improved mitochondrial oxidative function after normalization of 25OHD3 levels in 12 severely vitamin D3-deficient patients with chronic fatigue and myopathy (166). However, large interventional studies and clear evidence for usefulness of vitamin D3 treatment are still lacking. One cause of obvious reluctance to undertake larger interventional studies may be the ongoing debate and overall uncertainty about doses and possible side-effects of vitamin D3 treatment.
Timely diagnosis of underlying vitamin D3 deficiency or insufficiency and adequate treatment, even at the stage of unexplained chronic fatigue, is warranted. Measuring the blood levels of the precursor 25OHD3 is easy and cost-effective. In contrast, an elevated level of the active metabolite 1,25(OH)2D3 does not indicate vitamin D sufficiency, but might be an important clue to calcium deficiency, presumably associated with autoimmunity (2). Sufficiency is defined as 25OHD3 levels above 30-100 ng/ml (75-250 nmol/l). Levels of 10-30 ng/ml (25-75 nmol/l) indicate insufficiency, those below 10 ng/ml (25 nmol/l) clear-cut deficiency. Therapy is equally easy. Cholecalciferol can be applied orally, and continuous daily substitution is recommended. The therapeutic dose ranges from 4,000 to 10,000 IU (100-250 μg)/day. Higher doses of about 10,000 IU/day, and concurrent mineral, or other co-factor substitution are warranted in order to overcome eventual vitamin D3 resistance, such as in calcium deficiency. In contrast to drugs usually recommended for chronic inflammatory, autoimmune or malignant diseases, cholecalciferol is very inexpensive.
Early treatment of chronic fatigue and recurrent infections might prevent full-blown CFS/ME. Elevating 25OHD3 levels in early stages of diseases may ameliorate the course, and shorten the time needed for induction therapy, thus lowering total treatment costs. Deleterious side-effects, often life-threatening, of modern biological, cytostatic or immunosuppressive compounds may be avoided, or at least reduced. Incidence of relapse and treatment resistance may decrease, as well as the burden of disease, and therapy costs. However, usefulness and cost-effectiveness of vitamin D3 co-treatment needs further investigation from high-powered and carefully designed clinical studies.
Acknowledgements
I thank Gerhard RF Krueger MD PhD, UT – Houston Medical School, Houston, USA, for his support and advice in preparing this review.
Footnotes
-
Conflicts of Interest
None.
- Received September 9, 2013.
- Revision received November 4, 2013.
- Accepted November 6, 2013.
- Copyright © 2014 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
In this issue




{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Sufficient Supply of Vitamin D3 is Important for Proper Human Cell Functions and Stress Response
- Physical and Functional Epithelial Barriers are Enhanced by 1,25(OH)2D3
- Influence of 1,25(OH)2D3 on Antimicrobial Peptides (AMiPs)
- Auto/Xenophagy as an Important Cellular Rheostat and its Relation to 1,25(OH)2D3 and Cathelicidin
- Endo- and Phagocytosis are Enhanced by 1,25(OH)2D3 and Cathelicidin
- 1,25(OH)2D3 Induces Immune-tolerant Myeloid Dendritic Cells and Regulatory T-Cells
- Cathelicidin and NK cells in Innate and Adaptive Immunity
- Vitamin D Receptor and 1,25(OH)2D3 Are most Important for Invariant NK T-cells
- Intra-epithelial CD8αα TCRαβ Cells are Invariant NK T-cell-equivalent gut Mucosa Cells and Essential for Local Immune Balance
- Activated B-cells Express VDR, and B-cell/invariant NK T-cell Interactions Modulate Immune Responses
- Discussion
- Acknowledgements
- Footnotes
- References
- Figures & Data
- Info & Metrics