


Abstract
Aim: To generate and characterize a telomerase-immortalized human retinal microvascular endothelial cell (HREC) line. This cell line may be utilized as an in vitro model to study the molecular basis of several diseases of the human retina. Materials and Methods: Primary retinal neuronal cells were isolated and transfected with plasmid encoding full-length human telomerase reverse transcriptase (hTERT). Transfected cells were selected and characterized to determine telomerase activity, karyotype, proliferative capacity and functionality. Results: HREC-hTERT cells appear morphologically similar to primary endothelial cells and have an extended in vitro life-span. HREC-hTERT cells express the progenitor/stem cell marker nestin. They have active telomerase and a high proliferative capacity. These cells also maintain a diploid karyotype. The HREC-hTERT cells showed high colony-formation capacity and plating efficiency compared to the primary cells. These cells are capable of differentiation into neuronal and glial cell phenotypes and the differentiated cells express the astrocyte marker glial fibrillary acidic protein (GFAP) and the neuronal marker microtubule-associated protein-2 (MAP2), respectively. Conclusion: The in vitro life-span of human retinal neuronal endothelial cells can be extended by ectopic expression of hTERT without altering the genetic stability and functionality of these cells. These cells will be a valuable tool to further our understanding on the role of HRECs in the human blood-retinal-barrier and in angiogenesis and neovascularization.
Retinal edema, hemorrhage, ischemia, microaneurysms, and neovascularization characterize age-related macular degeneration (AMD) and diabetic retinopathy, two major ocular diseases that affect millions of people all over the world. AMD is the most common cause of irreversible blindness in older adults in the United States and other industrialized nations. Treatment of AMD is still an unmet need as there are no FDA approved therapies, except for anti-vascular endothelial growth factor (VEGF-A) therapy for individuals with late-stage disease. Furthermore, there are no approved treatments for the more common, dry form of AMD and other retinal disorders such as diabetic macular edema, retinal vein occlusion and diabetic retinopathy. One of the earliest indications of AMD and diabetic retinopathy is altered vascular permeability (1, 2); however, what causes changes in vascular permeability is still not completely understood, for lack of a better understanding of the inner blood–retina barrier (BRB) and the response of retinal microvasculature to changes in the microenvironment. The inner BRB is a complex system composed of highly specialized microvascular endothelial cells embedded in a basement membrane and surrounded by astrocytic end feet.
A wealth of information is available in the literature on the use of cultured retinal vascular endothelial cells from human and bovine sources (3-5). However, normal mammalian microvascular endothelial cells can be maintained in culture only for limited passages before they reach replicative senescence. This is a major problem in studying microvascular pathobiology. In addition, primary cells are difficult to work with due to variations in donor age, gender and the method of preservation of the tissue, most of which is eye bank tissues obtained from older donors. Due to the number of problems encountered while working with primary cells, most of the in vitro studies, including high-throughput screening for drug discovery, is done by using non relevant endothelial or cancerous cells, such as human umbilical vein endothelial cells (HUVECs) or BaF3 cells. Therefore, extending the life-span of endothelial cells without losing their primary features has enormous value for retina research.
Classically, large or small T-antigen of the Simian Virus strain (SV-40), papilloma virus (HPV) envelope proteins E6 and E7 (alone or in combination) and adenoviral proteins (AE1 and BE1) have been used to immortalize human and animal cells. However, in the process of by-passing the mortality check-points 1 and 2, the expression of the oncoproteins p53, retinoblastoma protein (pRb), or both is abrogated in resulting cell lines (6). The SV-40 large T-antigen binds to and inactivates the Rb and p53 in these cells, and as a result these cells do not respond to DNA damage (7) and have altered expression of cell-cycle proteins (8). The cells immortalized by E6/E7 do not express p53 protein and have inactive Rb, hence they have altered cell-cycle regulators and are immortal (6). Like any virally immortalized cells, the aforementioned cells are pluriploid and may not be appropriate models of certain cellular processes.
Primary antibodies used in these studies. The source of the various antibodies used and the dilutions used in western blot (WB) and immunofluorescence (IF) assays are indicated.
Since somatic cells undergo telomere shortening at each cell division, the strategy for the immortalization process involves activation of telomerase, the enzyme that maintains telomere length in stem cells and cancer cells (6, 9, 10). Another strategy to generate immortal cell lines is the introduction of ectopic expression of human telomerase reverse transcriptase (hTERT) into normal mammalian cells, which results in generation of cell lines with extended life-span (9). The advantage of constitutive hTERT activity is that it arrests telomere shortening and promotes mitosis. Several cell lines from different tissue origins [epithelial (11-13), stromal/fibroblast (7, 14, 15) and endothelial (16-19)] have been generated by ectopic expression of telomerase. The cell lines generated by this strategy have been shown to be diploid, very similar to their mortal counterparts, and to maintain normal functionality. Some studies have also reported the use of telomerase in combination with SV-40 large T-antigen (20-22) and HPV-16 E6 and E7 genes as immortalization strategies (23-25). However, these studies have also reported genetic instability in the immortalized cells (23). Recently, a bovine retinal vascular endothelial cell line was established with extended life-span by ectopic expression of telomerase (26). The purpose of this study was to establish a human retinal microvascular endothelial cell line with expanded life-span by ectopic expression of the human telomerase gene (hTERT). We also performed initial characterization of the cell line in order to utilize it for examining pathways implicated in disease processes of the retina.
Materials and Methods
All tissue samples were obtained in compliance with good clinical practice, with informed consent under Institutional Review Board (IRB) regulations, and also in accordance with the tenets of the Declaration of Helsinki.
Primary human retinal microvascular endothelial cell (HREC) isolation. HREC cells were isolated by modification of a previously-described protocol (27). Briefly, a neuro-retinal layer was isolated from the posterior segment of donor retina. The sample was removed and the retinal pigment epithelium (RPE) separated by gentle brushing with a sterile spatula and washing twice with sterile phosphate-buffered saline (PBS). The tissue was then minced and digested with 2 mg/ml collagenase type I for 30 min with constant agitation. The obtained cell suspension was washed several times with culture medium (EGM-2MV; Lonza, Allandale, NJ, USA) and then plated on 25-cm2 flasks or 6-well plates.
Cell culture. Primary HREC and HREC-hTERT cells were maintained at 37°C in 5% CO2 in culture media (EGM-2MV and NGM; Lonza, Allandale, NJ, USA). For progressive passages, cells were trypsinized with 0.05% trypisn-EDTA (Gibco, Carlsbad, CA, USA) and cultured in laminin-coated 60-mm dishes. The isolated primary HRECs can be progressively passaged for up to 6-8 passages and maintain their morphological and physiological properties.
Other cell types. ARPE19 cells (kind gift from Dr. Raghu Krishnamoorthy, UNTHSC, Fort Worth, USA) were cultured in DMEM containing 10% fetal bovine serum. The cells were maintained at 37°C in 5% CO2. For progressive passages, cells were trypsinized with 0.05% trypisn-EDTA (Gibco) and cultured in 60-mm dishes.

A: Phase-contrast image at indicated magnifications of HREC-hTERT cells in plastic culture dishes. B: Growth curve of HREC-hTERT cells and primary HREC cells. C: Population doubling time of HREC-hTERT (at passage12) cells was determined and compared with primary RPC cells (at passage 3) (Control). Data are reported as the mean of three independent experiments.
Lipofectamine-mediated cell transfection. Isolated HRECs from three different donors (age of donor: 76, 80 and 88 years) were used for transfection. Cells were seeded in a 6-well plate at a density of 50-60%. The following morning, they were transfected with pGRN145 plasmid (American Type Culture Collection (ATCC), Manassas, VA, USA) which encodes the full length hTERT. Escherichia coli containing the plasmid obtained from ATCC, were amplified in Luria Broth (LB) medium and plasmid DNA was isolated using the Maxiprep kit (Qiagen, Valencia, CA, USA) according to manufacturer's instructions. Lipofectamine-mediated transfection was performed according to the manufacturer's instructions. Transfected cells were selected using 200 μg/ml hygromycin B (Sigma Aldrich, St.Louis, MO, USA).
Immunocytochemistry. Approximately 15,000 cells were plated on glass coverslips (12 cm2; Fisher Scientific, Pittsburgh, PA, USA) and cultured in their respective media. When the cultures had stabilized, the coverslips were rinsed in phosphate-buffered saline (PBS), and fixed/permeabilized in methanol:acetone (1:1, 10 min at −20°C). After re-hydration in PBS (for 30 min) and distilled water washes (3×), the cells were blocked (overnight at 4°C) in PBS with 1% bovine serum albumin (BSA). The cells were then rinsed with PBS and distilled water (3×) and incubated at 4°C overnight with the primary antibody diluted in PBS (Table I). After rinsing in PBS containing Tween 20 (0.1%; 3×10 min), cells were incubated with secondary antibody at room temperature (RT, 1.5 h) and rinsed in PBS with Tween 20 (0.1%, 3×10 min). Alexa Fluor 594 nm goat anti-mouse, Alexa Fluor 594 nm goat-anti-rabbit and Alexa Fluor 594 nm donkey anti-goat (Molecular Probes/Invitrogen, San Diego, CA, USA) secondary antibodies were used at dilutions of 1:1000. The specimens were rinsed in PBS (3×10 min), distilled water (30 min), stained with 4’,6-diamino-2-phenylindole (DAPI) and mounted on glass slides (FluorSave, Calbiochem, La Jolla, CA, USA). Cells on coverslips stained with secondary antibody alone were used as negative controls, which showed virtually no fluorescence.
Image acquisition. Mounted specimens were examined on an Olympus Provis AX70 fluorescent microscope attached to an Olympus DP70 digital camera.
Western blot analysis. Cultured endothelial cells were treated with lysis buffer (2.5 ml 1M Tris buffer (pH=7.0), 1 g sodium dodecyl sulphate (SDS), and 2.5 g sucrose in 50 ml distilled water) for 5 min at RT. Genomic DNA was sheared by several passes through a 22-gauge needle, and samples were stored at −20°C. Bicinchoninic acid (BCA) protein assays (Pierce, Rockford, IL, USA) of lysates were performed to determine the protein concentration (and ensure equal loading of lanes). SDS-polyacrylamide gel electrophoresis (SDS-PAGE) was performed at RT, and 20 μg protein/lane was loaded, and the gel was run at 150 V in Tris/glycine as the running buffer. Protein bands were transferred onto nitrocellulose membranes (VWR International, Irving, TX, USA) by electro-blotting overnight (4°C) at 10 V in Tris/glycine buffer with 20% methanol, and transfer was confirmed with Ponceau Red (Sigma-Aldrich, St Louis, MO, USA) staining of the membranes. After de-staining in distilled water, membranes were incubated in blocking buffer (5% powdered milk and 1% BSA in PBS) for 1 h (RT). Membranes were then incubated with the primary antibody for 30 min (RT), then overnight (4°C) and, finally, the following morning for 30 min (RT). After rinsing in PBS containing 0.1% Tween-20 (3×10 min), the membranes were incubated with secondary antibody for 1 h (RT), rinsed in PBS with 0.1% Tween-20 (3×10 minutes), and developed (ECL Chemiluminescence, Amersham Biosciences, Little Chalfont, UK). HUVECs and retinal microvascular endothelial cell (RMVEC) lysates were used as a positive control for platelet endothelial cell adhesion molecule (PECAM) expression. Spontaneously immortalized retinal pigment epithelial cell (APRE19) lysates were used as a positive control for α-smooth muscle actin expression. Human primary astrocytes were used as a positive control for GFAP expression.
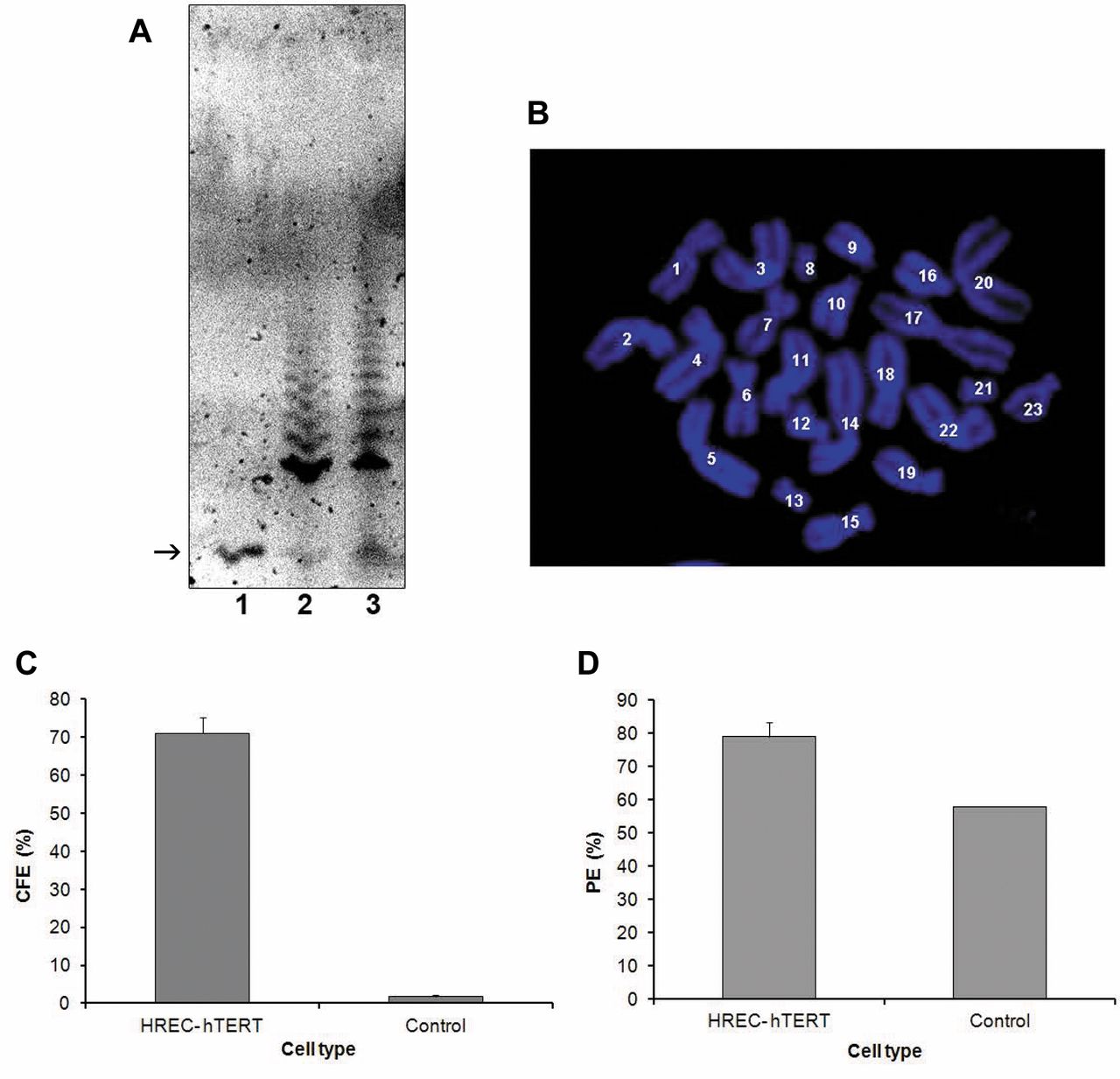
A: Telomerase activity. The TRAP assay was positive for telomerase activity at passage 12 for HREC-hTERT cells. There was no detectable telomerase activity in primary RPC cells. Arrow: The internal telomerase standard (ITAS) marker (internal control). Lane1: Primary RPC cells, lane 2: HREC-hTERT cells and lane 3: positive control (TSR8-TRAPeze kit). B: Karyotype analysis. In 30 cells examined at greater than 16 population doublings, karyotype analysis demonstrated 23 diploid chromosome pairs. C: Colony forming efficiency (CFE) of HREC-hTERT cells (at passage 12) was compared to that of primary cultures following low density seeding and counting the number of cells that resulted in colony formation. D: Plating efficiency (PE) of HREC-hTERT (at passage12) was also compared to that of primary cultures by determining the percentage of cells that plated down following trypsinization and re-seeding. Data are reported as the mean± standard deviation (SD) of three independent experiments.

Comparison of HREC-hTERT cells and ARPE19 cells by two-dimensional (2D) gel electrophoresis. Lysates from HREC-hTERT cells (at passage 15) (A) and ARPE19 cells (B) were analyzed by 2D gel electrophoresis. Circles denote differences in the two samples following Coomassie blue staining of the 2D gels. Black arrow indicates the internal loading control.
Determination of plating efficiency and population doubling time. Primary and HREC-hTERT cells were seeded at 100,000 cells in 6-well plates (2 wells/cell type). After 5 h, the number of cells that did not attach were counted to determine the actual number of cells that were seeded per well. Cells were then harvested and counted at 18, 24, 36 and 72 h. The ratio of the number of cells seeded to the final number of cells counted at different time points was used to determine the population doubling time.
Telomeric repeat amplification protocol (TRAP) assay. A TRAP assay was performed (TRAP-eze Telomerase Detection Kit; Chemicon, Temecula, CA, USA), and the TRAP activity was assessed in HREC-hTERT cells. Briefly, cell lysates from 100,000 cells underwent telomerase extension followed by PCR amplification and PAGE for gel-based telomerase detection. The gel was stained in buffer that contained ethidium bromide and imaged using an ECL machine (Amersham Biosciences). Lysis buffer was used as negative control.
Colony Forming Efficiency (CFE). CFE was calculated by seeding 500 cells/well into three wells of a 6-well plate. The number of floaters was counted the following day to determine the exact number of cells that had attached. After nine days of culture the cells were fixed in 10% trichloroacetic acid (TCA) at 4°C for 10 mins. The cells were then washed three times with distilled water. The colonies were stained using 0.4% sulforhodamine B (SRB) in 1% acetic acid for 10 min at RT. The cells were then washed four times with 1% acetic acid and allowed to air dry. The colonies were counted under a dissection microscope.
Karyotype analysis. Cells were cultured in 6-well (three wells/cell type) plates till they were 50% confluent. The cells were then treated with 10 μg/ml colcemid (Sigma-Aldrich) in culture medium overnight at 37°C in an incubator. Cells were passaged as described earlier using Trypsin-EDTA (Invitrogen, San Diego, CA, USA). The cell pellet was incubated in 500 μl of 0.75 M KCl (Sigma-Aldrich) at 37°C for not more than 25 min. The cells were then centrifuged at 4,000 rpm (Beckman Coulter Microfuge 22R, Brea, CA, USA), and KCl was removed. Freshly-prepared cold fixative (methanol:acetic acid, 1:1) was added and gently pipetted in order to break up the cell pellet. The cells were incubated at 20°C for 15 min, centrifuged at 4,000 rpm (Beckman Coulter) and the fixative was removed. Fresh fixative was once again added and cells were incubated at −20°C for another 15 min. Finally, the cells were placed onto slides, pre-cleaned with 100% ethanol and dried (Thermo Fisher, Pittsburgh, PA, USA), from a height of 8 inches. Slides were stained with DAPI (Molecular Probes/Invitrogen, San Diego, CA, USA) and the number of chromosomes was counted under a fluorescence microscope.
Two-dimensional gel electrophoresis. Two-dimensional gel electrophoresis was performed on equal amounts of HREC-hTERT and ARPE19 cell lysates followed by Coomassie blue staining (Kendrick labs, Madison, WI, USA).
RNA isolation and RT-PCR analysis. RNA was isolated using Trizol® reagent according to manufacturer's recommendations for cells cultured as a monolayer (Invitrogen). Trizol (1 ml/cm2 of cell monolayer) was added to the washed cell monolayer and the cells were detached from the flask using a cell scraper. The cell mixture was transferred to centrifuge tubes and incubated at 30°C for 5 min and then scraped. To this, 0.2 ml of chloroform was added and the closed tube was shaken vigorously by hand for 15 s, opened and incubated at 30°C for 2-3 min. The sample was then centrifuged at no more than 12,000 × g (2-8°C) for 15 min. The top aqueous layer was carefully removed and transferred to a fresh centrifuge tube and the RNA was precipitated with isopropanol (0.5 ml/ml of Trizol reagent used). The precipitated RNA was washed twice with ethanol and finally dissolved in diethylpyrocarbonate (DEPC) treated water. Primers pairs used for RT-PCR were for Nestin and beta tubulin III. RT-PCR was performed using Superscript® One-step RT-PCR system (Invitrogen).


Characterization of HREC-hTERT cells. A: Immunocytochemical analysis of (i) vWF (red), (ii) PECAM-1 (green), (iii) β-Actin (red), (iv) secondary antibody control. B: Western blot analysis was performed to determine the presence of marker proteins. Lane 1: HUVEC, lane 2: RMVEC, lane 3: HREC-hTERT and lane 4: ARPE cells. Western blot was developed using PECAM-1, α-SMA, GFAP and β-Actin antibodies.
Agarose gel electrophoresis. Agarose gel (1.2%) was prepared by heating agarose in Tris-acetic acid-diethyl tetra acetic acid (TAE) buffer. After cooling, ethidium bromide (6 μl in 100 ml of solution) was added to the mixture. The total sample obtained from RT-PCR was loaded onto the gel with 5 μl of bromophenol blue dye and imaging was performed using an imaging device (UVP Bioimaging system model EpiChemi3 Darkroom, Upland, CA, USA).
Results
Isolation of primary HRECs. We successfully isolated primary HRECs from several human donor tissues (n>12) of varying gender and age. The cells grew with typical cobblestone morphology (data not shown). It was possible to maintain the cells in culture for up to 6-7 passages, after which they would senesce.
Cell kinetics and morphology. From cells transfected from three different donors, HRECs from only one donor (76-year-old) proliferated past their normal in vitro life-span and overcame senescence. The cells were maintained in culture much longer (>passage 20) than the primary untransfected cells. The HREC-hTERT cells have cobblestone morphology, similar to primary HREC cells, which is typical of endothelial cells (Figure 1A). The growth curve shows that growth of primary (control) HRECs begin to plateau after passage 4, indicating a decrease and normalization of cell proliferation and eventually cell senescence (Figure 1B). In HREC-hTERT cells, there is a constant increase in cell number, indicating constant proliferation (Figure 1B). The time taken for a known population of cells to double was determined in HREC-hTERT cells that had undergone over 10 population doublings. The cell number did not change in 24 h. The population doubling of HREC-hTERT cells /population doubling of control ratio was 2.5 at 36 h (Figure 1C). The population doubling time of the hTERT cells was thus determined to be between 24 to 36 h. Primary HRECs have a doubling time of approximately 48 h (Figure 1C).
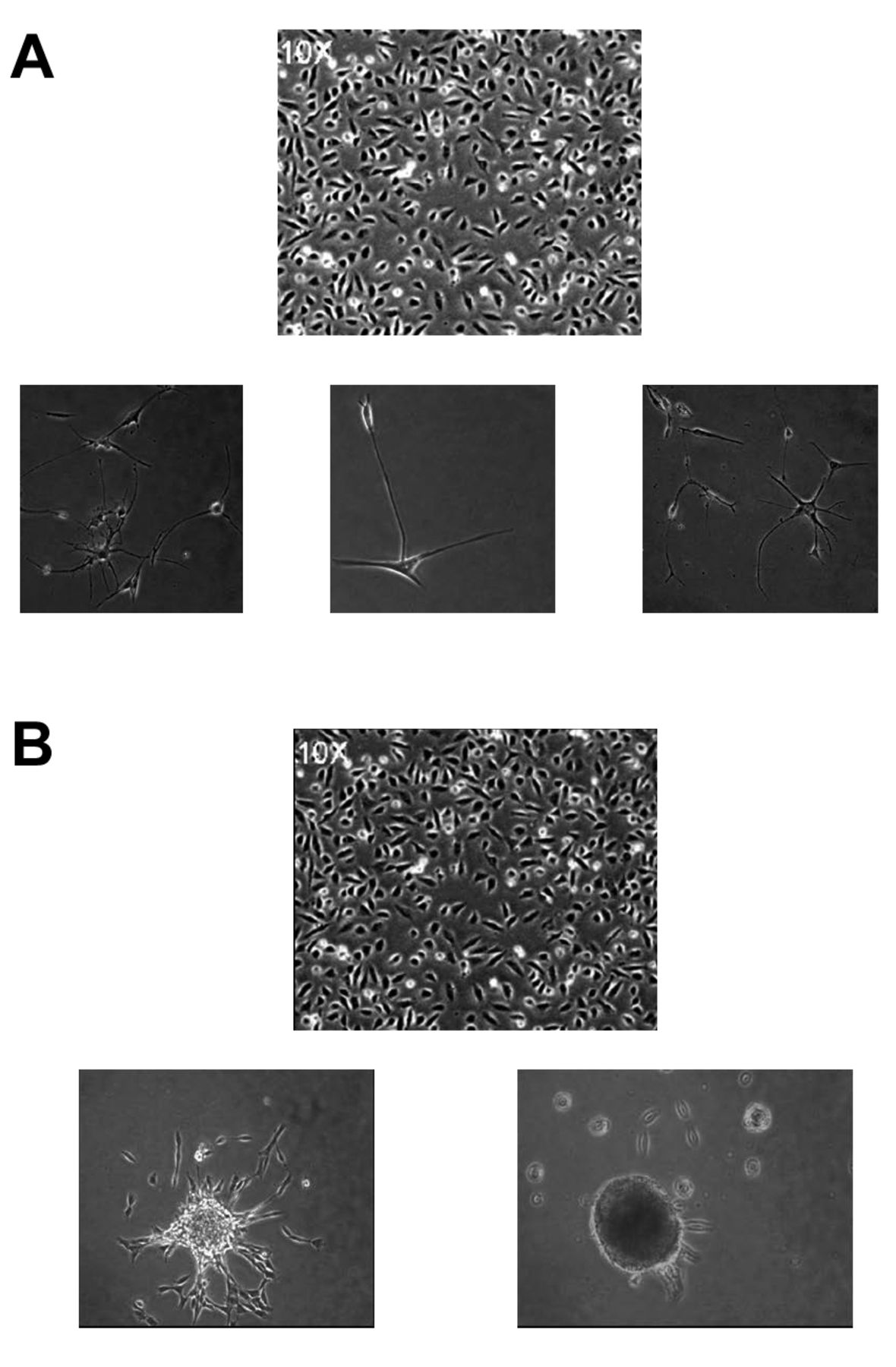
Differentiation into neuro-glial phenotypes. Morphology of HREC-hTERT cells in the presence of fibroblast growth factor (FGF) (A) and neurosphere formation by HREC-hTERT cells in NGM growth medium and on laminin IV, extracellular matrix (B).
Telomerase activity in HREC-hTERT cells. To determine the presence of telomerase activity in HRECs, TRAP assay was performed as described in the Materials and Methods section, using the TRAP-eze telomerase detection kit. Telomerase activity was indicated by the ladder of the amplified products. The HREC-hTERT cells were positive for telomerase activity as shown in Figure 2A, lane 2. There was no amplification and hence no telomerase activity in primary HRECs as can be seen in lane 1 of Figure 2A. The control sample TSR8 (lane 3) provided by the manufacturer, served as a positive control for telomerase activity.

Characterization of HREC-hTERT cells under differentiation conditions. Immunocytochemistry was used to determine the expression of GFAP (green) (A), nestin (red) (B) and MAP2 (red) (C).
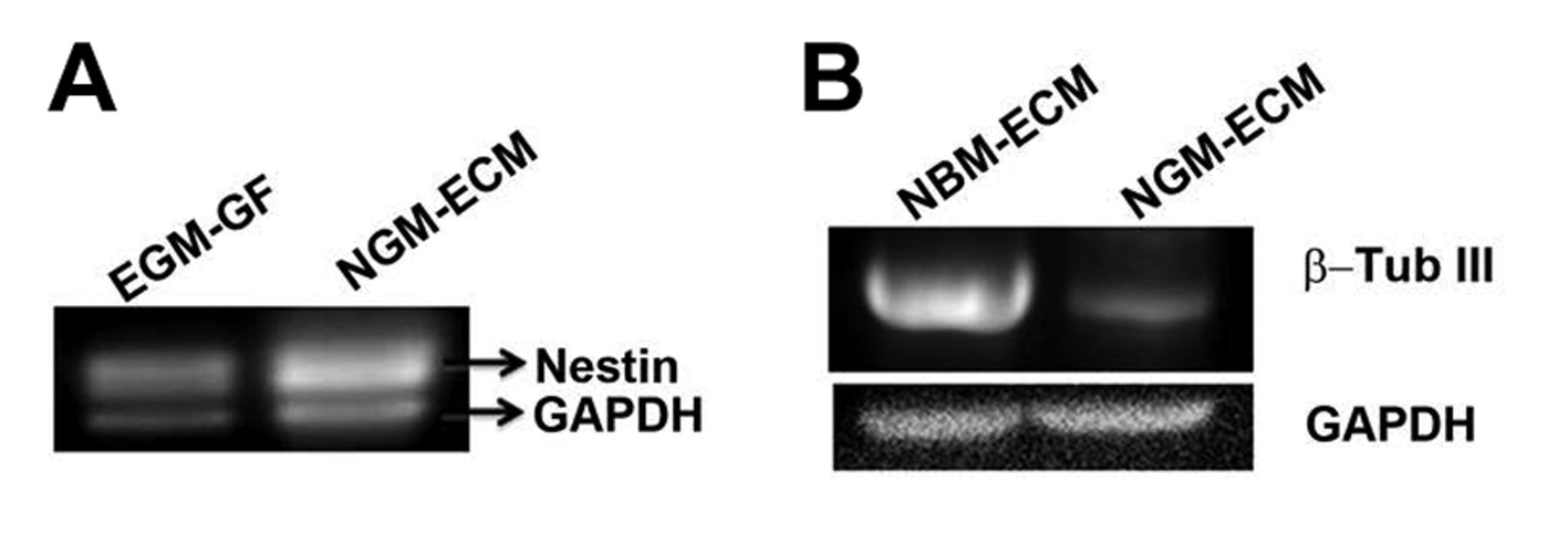
RT-PCR analysis to determine mRNA expression levels of neuronal marker β-tubulin III (β-Tub III) and progenitor/stem cell marker nestin in HREC-hTERT cells under different culture conditions. Endothelial growth medium-Growth factor (EGM-GF); Neuronal growth medium-Extracellular matrix (NGM-ECM); Neuronal basal medium-Extracellular matrix (NBM –ECM).
Karyotyping analysis of HREC-hTERT cells. The HREC-hTERT cells were arrested in metaphase and metaphase spreads were generated and stained with DAPI (Figure 2B). The number of chromosomes was then counted. The HREC-hTERT cells (at passage 13) were diploid, with a chromosome number of 46. Chromosomes from 30 cells were counted in order to confirm this.
Determination of CFE and plating efficiency in HREC-hTERT cells. The HREC-hTERT cells, plated at a very low density, gave rise to colonies in 10 days, and 71% of the plated cells resulted in colonies. Primary HREC cells at passage 1 were used as a negative control, where fewer than 2% of the proliferating population of cells resulted in colonies (Figure 2C). The HREC-hTERT cells had a plating efficiency of 76%, while the primary HRECs had a plating efficiency of 58% (Figure 2D).
Two-dimensional (2D) gel electrophoresis. In order to eliminate the possibility that the HREC-hTERT cells contained any RPE cells, two-dimensional gel analysis was performed. Cell lysates from HREC-hTERT and ARPE19 cells were visually compared following Coomassie blue staining of the gels. As shown in Figure 3, 15 different spots were identified that are indicative of proteins that are differentially expressed in the two samples.
Determination of marker proteins. The HREC-hTERT cells did not express endothelial cell markers von Willebrand factor (vW) (Figure 4A panel i) or PECAM (Figure 4A, panel ii) as determined by indirect immunofluorescence analysis and western blot analysis (Figure 4B) at passages 12-14. The cells did express β-actin (Figure 4A, panel iii). They did not stain positive for α-smooth muscle actin which is expressed in RPE cells, or for GFAP, which is a glial/astrocyte marker (Figure 4B). The antibody controls showed virtually no fluorescence (Figure 4A, panel iv). The cells were negative for 14-3-3 sigma, which is an epithelial cell marker, as determined by western blot analysis (data not shown).
Differentiation of HREC-hTERT cells. In the presence of culture medium (EGM-2MV and NGM), the HREC-hTERT cells were highly proliferative. When these cells were plated on laminin IV and the culture medium was NGM, they continued to proliferate but eventually formed neurospheres that detached from the flask and continue to survive in suspension (Figure 5). The cells from the neurospheres plated down on laminin IV-coated surfaces in the presence of NBM (absence of growth factors) and differentiated into a neuronal and glial cell mixed population. These cells expressed neuronal marker MAP-2 and astrocyte marker GFAP (Figure 6). The differentiated cells also expressed β-tubulin III, a neuronal marker, as shown by RT-PCR (Figure 7). The proliferative cell population of HREC-TERT cells expressed nestin, as shown by indirect immunofluorescence and RT-PCR analyses (Figures 6 and 7).
Discussion
Studies utilizing primary cells have their restrictions due to their limited in vitro proliferative capacity. Our studies have shown that primary cells can be cultured in vitro successfully up to passage 8 depending on the condition of tissue obtained from the eye banks. The other problem encountered during culture of primary HRECs is cross-contamination with RPE cells, a highly proliferative cell type, which could result in misinterpretation of data. Our study reports the development of a telomerase-immortalized human retinal progenitor cell line by ectopic expression of hTERT alone. Telomerase overexpression, as reported for several other endothelial and epithelial cell lines, maintains normal endothelial phenotype while exhibiting genetic stability. Our studies report the development of the very first telomerase immortalized human retinal progenitor cell line, which can differentiate into neuro-glial cell types that reside in the neuroretinal layer of the human retina.
A number of virally-transformed cell lines that have been generated are tetraploid (28, 29) but telomerase immortalized cells have been reported to maintain normal ploidy (30-32). We show that HRECs can have an extended in vitro life-span (passage >22) when transfected with hTERT. However, only HRECs from one donor resulted in a cell line with extended life-span, suggesting that telomerase immortalization may be dependent on the condition of the donor tissue. Another important fact to note is that we utilized lipofectamine-mediated transfection vs. the traditional and most commonly used retroviral strategy. The HREC-hTERT cells maintain normal epithelial morphology but appear smaller under phase-contrast microscopy. These cells are highly proliferative compared to the primary cells but maintain their diploid karyotype. These cells need to be characterized and karyotyped periodically, as the population doubling time and passage increases, in order to guarantee that the genetic stability and cell phenotype has not altered.
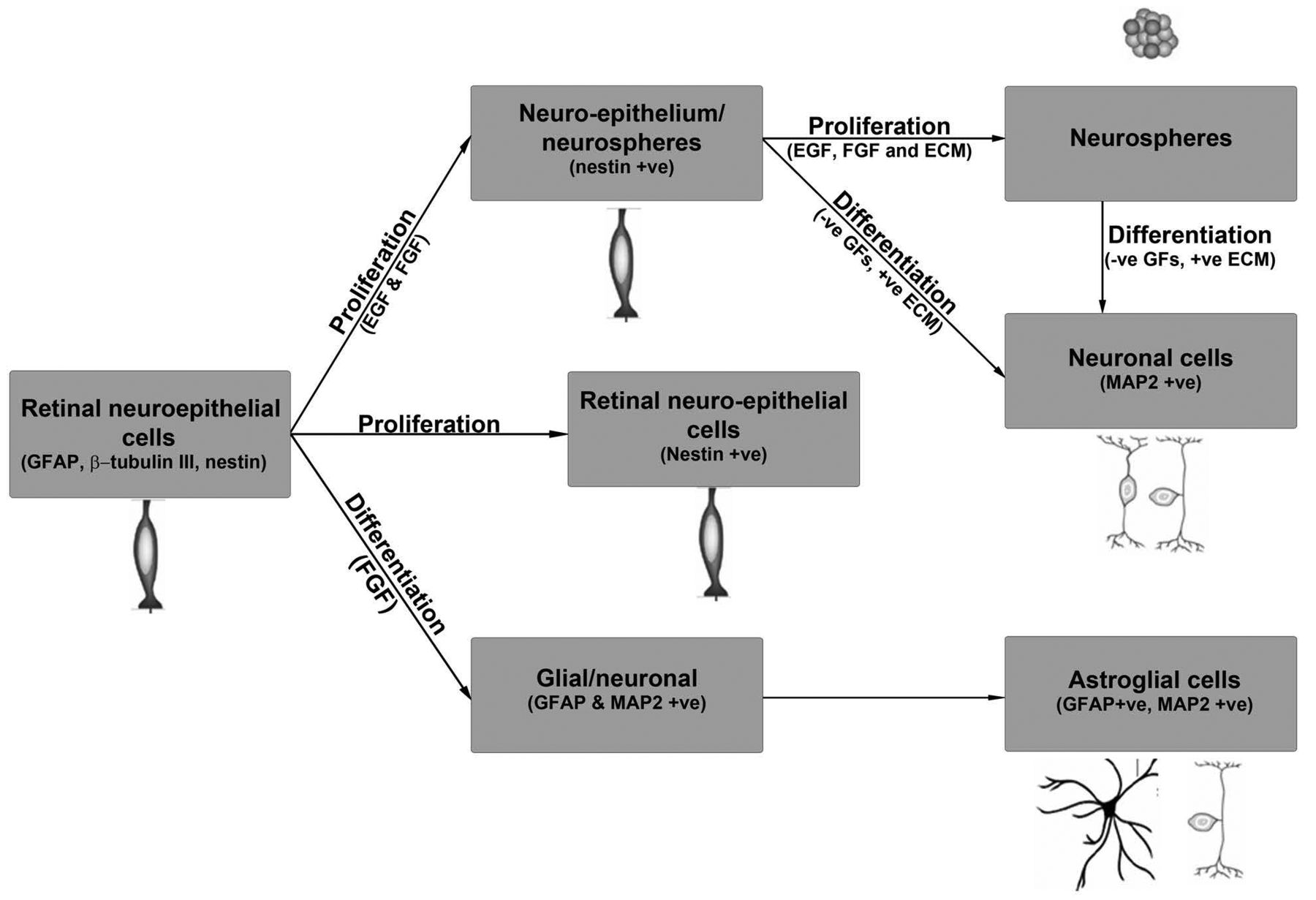
Illustration of proliferation and differentiation of HREC-hTERT under different culture conditions. All abbreviations are explained in the text.
The data reported in our studies are from HREC-hTERT cells between passages 12 and 15. The cells express endothelial cell markers as shown by immunofluorescence analysis and functionality of these cells was studied utilizing an in vitro angiogenesis/tube formation assay (data not shown). However, it is interesting to note that the primary HRECs form capillary-like structures in vitro at 12-24 h, whereas the HREC-hTERT cells take longer (>24 h) to form these structures. The structures, however, can be maintained in culture for long periods of time. In order to eliminate the possibility that there was a cross contaminating population of RPE cells, the HREC-hTERT cells were compared with the well-established ARPE 19 cells by two-dimensional gel analysis, showing differential expression of proteins in the patterns between the two samples.
The HREC-hTERT cells have high proliferative capacity up until the passage that we have characterized them (passage 22). Long-term in vitro culture and characterization may be required to establish if these cells are indeed immortal or they have a limited life-span. Differentiation of these cells into one specific and uniform phenotype also requires further studies on providing them with the right combination of growth factors and extracellular matrix.
In summary, we have developed a human retinal microvascular endothelial cell line with expanded life-span by ectopic expression of the human gene hTERT. We have also shown that these cells maintain normal morphology and are highly proliferative as compared to the primary cells. These cells have the ability to differentiate into different phenotypes depending on the growth factors and growth conditions, as illustrated in Figure 8. The availability of these cells allows us to conduct extensive studies to determine their role in maintaining the human BRB and understanding the physiological process involved in forming new blood vessels from pre-existing vessels in angiogenesis and neovascularization.
- Received June 20, 2013.
- Revision received July 12, 2013.
- Accepted July 16, 2013.
- Copyright © 2013 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}