


Abstract
Aim: The present study adds scientific support to the growing debate regarding the superiority of radiolabeled bombesin-based antagonist peptides over agonists for molecular imaging and therapy of human tumors overexpressing the gastrin-releasing peptide receptor (GRPR) and describes a detailed in vitro and in vivo comparison of 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 agonist and 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt antagonist ligands. Materials and Methods: Conjugates were synthesized by solid-phase peptide synthesis, purified by reversed-phase high-performance liquid chromatography, and characterized by electrospray ionization-mass spectroscopy. The conjugates were radiolabeled with 64Cu. Results: In vitro and in vivo data support the hypothesis for targeting of the GRPR by these tracer molecules. Maximum-intensity micro Positron Emission Tomography (microPET) imaging studies show the agonist ligand to provide high-quality, high-contrast images with very impressive tumor uptake and background clearance, with virtually no residual gastrointestinal or renal-urinary radioactivity. Conclusion: Based on microPET imaging experiments, we conclude the agonist peptide ligand to be a superior molecular imaging agent for targeting GRPR.
Peptide receptor-targeted radionuclide therapy is a method of site-directed radiotherapy that has been effectively used to specifically target human cancer that expresses a cognate receptor-subtype in very high numbers. Ideally, the procedure allows only the primary or metastatic disease to be targeted and is minimally invasive, with little radiation damage to normal, collateral tissues (1-11). Successful targeting of somatostatin receptor-positive tumors by receptor-specific diagnostic radiopharmaceuticals, has pioneered efforts to develop new biologically active targeting vectors that have high affinity and selectivity for human cancer cells (12, 13).
Prostate cancer is one of the most commonly diagnosed types of cancer in men and the second leading cause of cancer-related deaths in the United States. It accounts for about one in four newly diagnosed cases of cancer each year among U.S. men. An estimated 241,740 new cases of prostate cancer will be diagnosed in 2012, resulting in 28,170 deaths (14). Gastrin-releasing peptide receptor (GRPR) is a G-protein-coupled receptor highly expressed in prostate cancer cells, but also known to be expressed in high numbers on a variety of solid tumor types including breast, lung, and pancreatic cancer (7, 15). Markwalder and Reubi (8) have previously demonstrated that GRPR expression in primary prostatic invasive carcinoma was present in 100% of tested tissues, and in 83% of these cases, GRPR expression was determined to be high or very high (1,000 dpm/mg tissue). Furthermore, Sun and co-workers (16) also found that in 90% of patients identified with prostate cancer, GRPR expression was prevalent. These findings and other studies offer impetus for the development of GRPR-specific diagnostic and therapeutic radiopharmaceuticals targeting primary and metastatic prostate cancer.
Bombesin, a 14-amino acid amphibian homolog of mammalian gastrin-releasing peptide, binds to the GRPR with very high affinity and specificity. Several researchers around the globe have continued to improve the diagnostic and therapeutic capability of radiolabeled bombesin analogs towards GRPR-positive tumors, by using different radiometals, as well as diverse bifunctional chelators. In recent years, our group and many others have investigated copper-64 (64Cu) [t1/2=12.7 h; Eβ+max=0.65 MeV (17.9%); Eβ−max=0.57 MeV (39%); 43.1% electron capture] radionuclide as a promising isotope for site-directed positron-emission tomography (PET) (17-19). The half-life for 64Cu is sufficiently long for drug preparation, quality control, drug incorporation, circulation, and patient imaging/therapy (20-22). However, widespread usage of 64Cu radiometal as a diagnostic tool has been limited because of transmetallation reactions in vivo with serum proteins, namely, superoxide dismutase, found in blood and liver tissue.
Until now, the handful of radiolabeled bombesin analogs that have been successfully used for molecular imaging and therapy of human tumors overexpressing the GRPR have primarily been agonist-based constructs. Agonists have been preferred over antagonists because they become internalized upon binding to the receptor, demonstrating a vital mechanism for accumulation and retention of the radiolabeled conjugate within the targeted cells (23-25), which can be a prerequisite for optimal visualization and/or treatment of disease. Recently, Maecke et al. (26) have shown in tumor tissue from animal models that high-affinity somatostatin (sst) receptor antagonists, led to equal or better in vivo results in terms of uptake and retention than the corresponding radiolabeled agonist. The superior tumor uptake was explained by a higher number of binding sites recognized by antagonists in comparison to the agonist ligands. This observation has been described for each sst2- and sst3-selective somatostatin analog, which suggests that such a change of archetype may be legitimate for more than one particular G-protein–coupled receptors, as these radiolabeled constructs bind to distinct receptor subtypes. However Wadas et al. (27) also compared the antagonist 64Cu-CB-TE2A-sst2-ANT to the agonist 64Cu-CB-TE2A-tyrosine3-octreotate (64Cu-CB-TE2A-Y3-TATE) in commonly used, rat pancreatic AR42J cells. Interestingly, they did not find the superiority of the antagonist, despite the fact that a 14-fold higher number of binding sites for the antagonist was found. These contradictory findings indicate that the in vitro and in vivo properties of radiolabeled G-protein–coupled receptor antagonists vs. agonists are not yet distinguished or fully understood.
Recently, Abiraj and co-workers described in detail a new antagonist ligand framework of the general structure (PEG4-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2 [AR]), which when radiolabeled with either 99mTc, 111In, 68Ga, or 64Cu, showed favorable in vitro and in vivo effects (including molecular imaging investigations) for targeting GRPR-positive prostate carcinomas. The high-quality, high-contrast images produced using 64Cu-CB-TE2A-AR targeting vector led this group to begin clinical investigations for molecular imaging of GRPR-positive neoplastic tissues for patients presenting with primary prostate cancer (28).
In the present study we describe a detailed comparison of agonist and antagonist GRPR-targeting ligands in the form of [64Cu-NODAGA-6-Ahx-BBN(7-14)NH2] and [64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt] (Figure 1), where NODAGA is 1,4,7-triazacyclononane,1-glutaric acid-4,7-acetic acid and 6-Ahx is the pharmacokinetic modifier 6-aminohexanoic acid, BBN is bombesin, BBN(7-14)NH2 is Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2 (agonist) and DPhe6-BBN(6-13)NHEt is DPhe6-Gln-Trp-Ala-Val-Gly-His-Leu-NHEt (antagonist), respectively. Our newly constructed peptide-targeting vectors have high affinity for GRPRs localized on the surface of human prostate cancer. The PC-3 tumor cell line is known to express the GRPR in very high numbers and was therefore chosen as a model for targeting GRPR-expressing tumors for this study. Herein, we describe thorough in vitro and in vivo investigations including microPET molecular imaging studies for each of the new targeting vectors.
Materials and Methods
General. Rink amide methylbenzhydrylalanine (MBHA) and [3({ethyl-Fmoc-amino}methyl)indol-1-yl]acetyl (AM) resins for synthesis of the bombesin agonist and antagonist peptides were purchased from EMD Biosciences (San Diego, CA, USA). Fmoc-amino acids and coupling reagents were purchased from either EMD Biosciences or Advanced Chemtech (Louisville, KY, USA). NODAGA(tBu3) was purchased from CheMatech (Dijon, France). All other reagents/solvents were purchased from Fisher Scientific (Pittsburgh, PA, USA), Sigma-Aldrich Chemical Company (St. Louis, MO, USA) or ACROS Organics (Geel, Belgium) and were used without further purification. 125I-Tyr4-BBN was purchased from Perkin Elmer (Waltham, MA, USA). Copper radionuclide in the form of 64CuCl2 in 0.1 M HCl solution was purchased from the University of Wisconsin-Madison Medical Physics Department, USA. The peptide conjugates and their metallated complexes were purified using reversed-phase high-performance liquid chromatography (RP-HPLC) performed on an SCL-10A HPLC system (Shimadzu, Kyoto, Japan) employing a binary gradient system [solvent A=99.9% (DI) water with 0.1% trifluoroacetic acid (TFA); solvent B=acetonitrile containing 0.1% TFA]. Samples were detected using an in-line Shimadzu SPD-10A absorption detector (λ=280 nm), as well as an in-line EG&G Ortec NaI solid crystal scintillation detector (EG&G, Salem, MA, USA). EZStart software (7.3; Shimadzu) was used for data acquisition of both signals. A semipreparative C-18 reversed-phase column (Phenomenex, Belmont, CA, USA) maintained at 34°C with an Eppendorf CH-50 column heater was used for purification of crude peptides, while an analytical C-18 reversed-phase column (Phenomenex, Belmont, CA, USA), maintained at 34°C, was used to achieve purification of NODAGA-6-Ahx-BBN agonist and antagonist peptides.
Peptide synthesis and purification. NODAGA-6-Ahx-BBN(7-14)NH2 and NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt conjugates were synthesized on a Liberty automatic microwave peptide synthesizer employing traditional Fmoc chemistry. The sequential addition of amino acids was accomplished by reacting O-benotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro phosphate-activated carboxyl groups on the reactant with the N-terminal amino group on the growing peptide, anchored via the C-terminus to the resin. The peptide products were cleaved by a standard procedure employing a cocktail of 1,2 ethanedithiol, water, triisopropylsilane, and TFA (2.5:2.5:1:94), followed by precipitation into methyl t-butyl ether. The crude peptides were purified by RP-HPLC using a semipreparative C-18 reversed-phase column (250×10 mm, 10 μm). The mobile phase solvent composition was changed from 75% A:25% B to 65% A:35% B over a 15 min gradient at a flow rate of 5 ml/min to achieve separation. The column was flushed with a solvent composition of 5% A:95% B and then re-equilibrated to the original gradient solvent composition. Solvents were removed using a CentriVap system (Labconco, Kansas City, MO, USA). Electrospray ionization–mass spectrometry (ESI-MS) (MU-Department of Chemistry, Columbia, MO, USA) was employed to characterize all of the non-metallated and natural copper metallated conjugates.
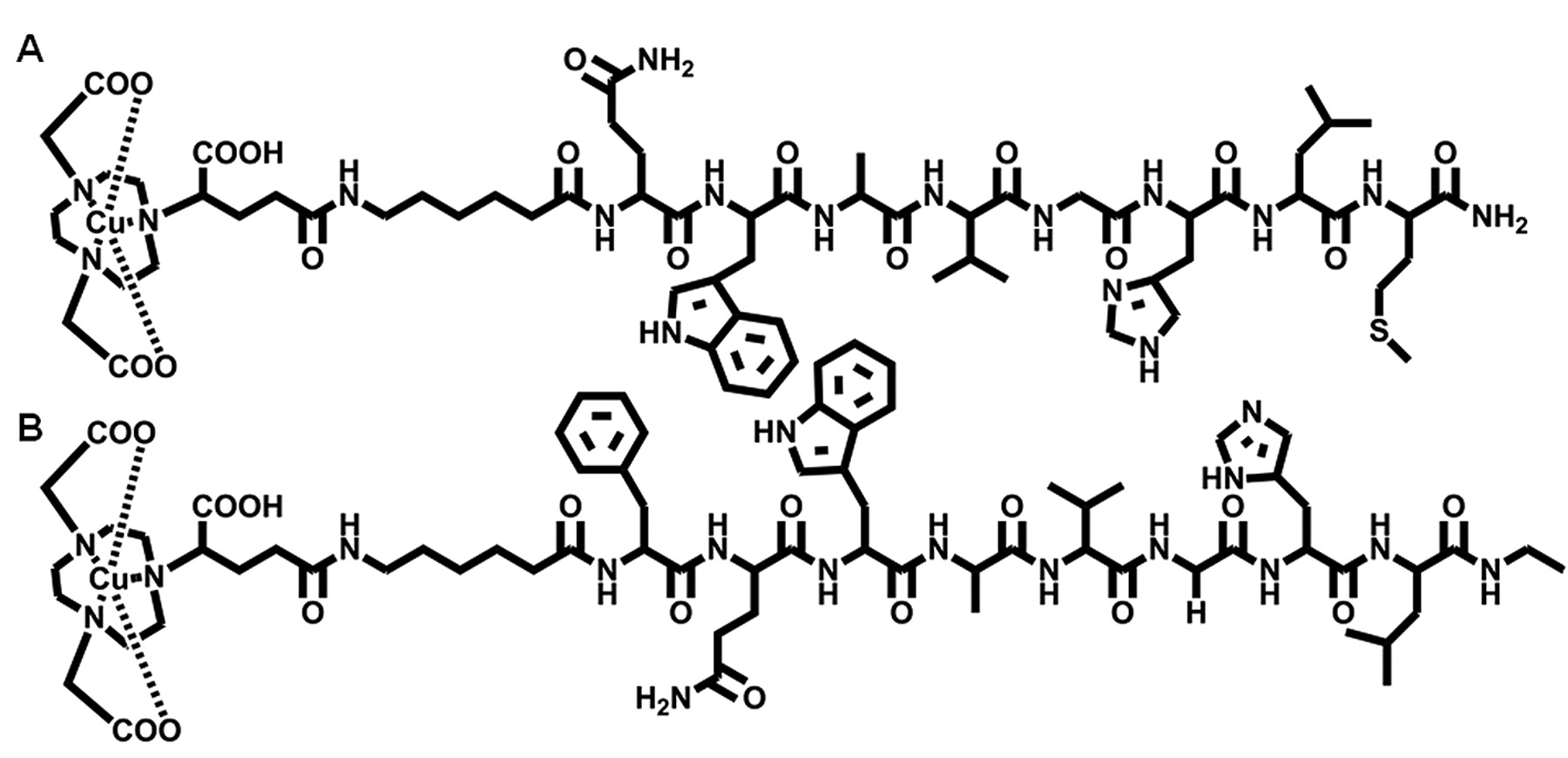
Representative structures of agonist and antagonist Gastrin Releasing Peptide Receptor (GRPR)-targeting ligands in the form of 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 (A) and 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt (B).
natCu and 64Cu labeling. natCu-NODAGA-6-Ahx-BBN(7-14)NH2 and natCu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt conjugates were synthesized by the addition of natCuCl2•2H2O in 0.05 N HCl (90 nmol) to a plastic tube containing purified peptide conjugate (80 nmol) and 0.4 M ammonium acetate (250 μl). The pH of the reaction mixture was adjusted to ~7 by the addition of 1% NaOH and then incubated for 1 h at 70°C. Ten millimolar diethylenetriaminepenta-acetic acid (DTPA) solution (50 μl) was added to scavenge unbound metal. Metallated conjugates were purified by RP-HPLC and analyzed by ESI-MS. The pure product was obtained as a white powder. 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 and 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt conjugates were synthesized according to a similar procedure by adding 64CuCl2•2H2O in 0.05 N HCl (9.15×1018 Bq/mol) to a plastic tube containing purified peptide conjugate (25 μg) and 0.4 M ammonium acetate (250 μl). The pH of the reaction mixture was adjusted to ~7 by the addition of 1% NaOH and then incubated for 1 h at 70°C. Ten millimolar DTPA solution (50 μl) were added to scavenge unbound radiometal. Finally, the radiolabeled conjugates were purified by RP-HPLC and collected into 1 mg/ml bovine serum albumin (BSA) (100 μl) and ascorbic acid (10 mg) stabilizing agent, prior to all in vitro/in vivo investigations. Acetonitrile was removed under a steady stream of nitrogen.
Serum stability. Upon RP-HPLC purification, the 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 and 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt conjugates were added to 500 μl of human serum albumin (HSA) (1 g/ml) in a small sample vial. Serum samples (pH 7.0±0.5) were incubated (at 37°C in a 5% CO2-enriched atmosphere) for 0, 1, 4, and 18 h. At each time point, the amount of 64Cu dissociation from the NODAGA ligand was assessed by RP-HPLC after serum proteins were removed via filtration through a 0.45 μm Millex-HV syringe filter (Millipore). Human Serum Albumin (HSA)-associated radioactivity was also evaluated by counting the radioactivity prior to loading and after elution of the filter.
In vitro cell binding affinity studies. A competitive displacement binding assay was used to assess the binding affinity of non-metallated and natural copper metallated conjugates of both the agonist and antagonist constructs for the GRPR using PC-3 human prostate cancer cells and 125I-Tyr4-BBN (Perkin-Elmer, Waltham, MA, USA) as the displacement radioligand. Approximately 3×106 PC-3 cells in Dulbecco's Modified Eagle Medium: Nutrient Mixture (D-MEM/F12K) containing 0.01 M MEM and 2% (BSA), pH=5.5, were incubated with 20,000 counts per min of 125I-Tyr4-BBN (2.7×10−11 mol, 3.18×1017 Bq/mol) and increasing concentrations of metallated and nonmetallated agonist and antagonist conjugates at 37°C (5% CO2-enriched atmosphere) for 1 h. After incubation, the reaction medium was aspirated and the cells were washed four times with cold medium. Cell-associated radioactivity was determined by counting the washed cells in a multiwell gamma counting system (Laboratory Technologies, INC., Maple Park, IL, USA). The percentage of 125I-Tyr4-BBN bound to the cells was plotted versus the concentration of metallated and nonmetallated conjugates to determine the respective 50% inhibitory concentration (IC50) values. The study was carried out in triplicate and the final IC50 was calculated by averaging the three experiments.
In vitro internalization and externalization assays. Internalization assays were determined in triplicate by incubating 3×104 PC-3 cells (in D-MEM/F12K media containing 0.01 M MEM and 2% BSA, pH=5.5) in the presence of 20,000 counts per min of both 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 and 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt conjugates (3.03×10−13 mol, 3.64×1019 Bq/mol) (at 37°C, in a 5% CO2-enriched atmosphere). At 10, 20, 30, 45, 60, 90 and 120 min post-incubation, the cells were aspirated, washed with fresh medium and acetic acid/saline (pH=2.5, 4°C) to remove surface-bound radioactivity, and counted on a multiwell gamma counter. Internalization was calculated relative to the total amount of activity added to the sample plate. Externalization assays were determined in triplicate after an initial 45 min internalization period in PC-3 cells. Incubation was interrupted by aspiration of cell media and washing with fresh medium. The cells were resuspended in fresh media and incubated a second time (at 37°C, in a 5% CO2-enriched atmosphere). At 0, 20, 40, 60, 90 and 120 min post-incubation, the cells were washed with medium and acetic acid/saline (pH=2.5, 4°C) and counted on a multiwell gamma counter to determine the amount of retained radioactivity.
Pharmacokinetic studies. All animal studies were conducted in compliance with the highest standards of care, as outlined in National Institute of Health's Guide for the Care and Use of Laboratory Animals and the Policy and Procedures for Animal Research at the Harry S. Truman Memorial Veterans' Hospital. Female CF-1 (23.23-28.6g) and Institute of Cancer Research (ICR) severely combined immunodeficient (SCID) female mice (4-5 weeks of age and 15.96-25.46g) were supplied from Taconic Farms (Germantown, NY, USA). The mice were housed at five animals per cage in sterile microisolator cages in a temperature- and humidity-controlled room with a 12 h light/12 h dark schedule. The animals were fed autoclaved rodent chow (Ralston Purina 300 Company, St. Louis, MO, USA) and water ad libitum. SCID mice were anesthetized for injections with isoflurane (Baxter Healthcare Corp., Deerfield, IL, USA) at a rate of 2.5% with 0.4 l oxygen through a non-rebreathing anesthesia vaporizer. PC-3 cells were injected subcutaneously into the bilateral flanks of the rodents, with ~5×106 PC-3 cells in a suspension of 100 μl normal sterile saline per injection site. Tumors were allowed to grow 2-3 weeks post-inoculation, developing tumors ranging in mass from 0.02 to 0.26 g. Biodistribution studies were performed in CF-1 and SCID mice after tail vein injection with ~20 μCi of the conjugate in 100 μl of isotonic saline. CF-1 mice were humanely euthanized at 1 h post-injection (p.i.) for the agonist ligand, whereas at 0.25 min and 1 h p.i. for the antagonist. Similarly, SCID mice were humanely euthanized at 1, 4, and 24 h p.i. for the agonist and 0.25 min, 1, 4, and 24 h p.i. for the antagonist. Tissues and organs were excised from the animals and were weighed, along with urine, and the associated radioactivity was counted in a well counter comprising a NaI(Tl) scintillation detector (ORTEC, Oak Ridge National Laboratory, Oak Ridge, TN, USA). The percentage injected dose (%ID) and the percentage injected dose per gram (%ID/g) for each organ or tissue were calculated. The %ID in whole blood was estimated assuming a whole blood volume of 6.5% of the total body weight. Urine activity was reported as %ID and consisted of radioactivity in the urine, bladder, and cage paper. The %ID/g of tumor tissue is reported as the average and standard deviation of the two individual bilateral xenografts.
MicroPET/micro-computed tomography (CT) imaging studies. Maximum intensity microPET coronal images were obtained on a Siemens INVEON small-animal, dedicated PET unit (Siemens, Nashville, TN, USA). The unit has a transverse field of view (FOV) of 10 cm and an axial length of 12.7 cm. The scanner was operated in a three-dimensional (3D) volume imaging acquisition mode. Mice bearing xenografted human prostate PC-3 tumors were administered 600-700 μCi of 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 and 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt conjugate in 100 μl of isotonic saline via tail vein injection. At 15 h p.i., the mice were humanely euthanized by CO2 administration and were laser-aligned at the center of the scanner FOV for subsequent imaging.
Image reconstruction was obtained with an ordered subset expectation maximization (OSEM) algorithm with 3D resolution recovery and the absence of tissue attenuation correction. The microPET data were filtered with a Gaussian full width at half maximum filter. Additionally, the micro-computed tomography (microCT) coronal images were also obtained on the Siemens INVEON small-animal CT unit, immediately after microPET for the purpose of anatomic/molecular data fusion. The microCT images were acquired for ~8 min, and concurrent image reconstruction was achieved using a conebeam (Feldkamp) filtered backprojection algorithm. The reconstructed (raw) microPET datasets (with a matrix size of 512×512×159) were imported into the INVEON Research Workstation software for subsequent image fusion with the microCT image data and 3D visualization.
Results
Agonist and antagonist targeting probes in the forms of NODAGA-6-Ahx-BBN(7-14)NH2 and NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt were synthesized and compared in detail to enhance the growing discussion regarding the superiority of agonists vs. antagonists as potential targeting vectors for the GRPR. Both of the conjugates were synthesized by solid-phase peptide synthesis utilizing traditional Fmoc chemistry with approximately 30% yield after RP-HPLC purification. The agonist [BBN(7-14)NH2] construct has the general structure Q-W-A-V-G-H-L-M-NH2, whereas the antagonist [DPhe6-BBN(6-13)NHEt] construct consists of the amino acid sequence DPhe6-Q-W-A-V-G-H-L-NHEt. In the antagonist construct, D-Phe was placed in the 6 position, as some recent reports suggest that the presence of a D- or an unnatural amino acid in the peptide sequence increases the resistance to proteolytic degradation (29, 30). Positioning of leucine at position 13 of the peptide sequence conferred antagonist behaviour onto the conjugate (31). Our laboratory has reported upon a very recent study on similar antagonist conjugates where we used dianionic NOTA chelator and observed a considerable degree of collateral radioactivity in the liver and abdominal region of microPET images (32). We propose that a charged ligand–metal complex at physiological pH might facilitate urinary excretion, reducing the amount of collateral radioactivity in normal surrounding tissues. Previous reports from our laboratory (10, 32, 33) suggest that NOTA complexing agent provides a high degree of thermodynamic stability, kinetic inertness, and very high in vivo stability to the 64Cu-chelate complex. For this study, we chose the tri-acetate version of NOTA, NODAGA, as a bifunctional chelating agent without compromising the in vivo stability of the conjugate. 6-aminohexanoic acid (6-Ahx) was used as a pharmacokinetic modifier for both the agonist and the antagonist targeting vectors. Metallation of the NODAGA-6-Ahx-BBN(7-14)NH2 and NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt conjugates was performed with natural copper (natCuCl2) and radioactive copper (64CuCl2). The radioactive copper conjugates were obtained in greater than 90% radiochemical yield as single products. All of the natCu and 64Cu conjugates were purified by RP-HPLC. The ESI-MS analyses of both the non-metallated and natural copper metallated peptide conjugates were consistent with the molecular weights that were calculated (Table I). Both of the 64Cu metallated conjugates demonstrated in vitro stability in excess of 24 h as monitored by RP-HPLC, with minimal observable degradation or transmetallation to serum proteins when challenged with HSA.
Electrospray mass spectrometry values for the non-metallated and metallated conjugates.
In vitro competitive cell binding assays of NODAGA-6-Ahx-BBN(7-14)NH2 and NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt conjugates and their natCu metallated constructs were performed with 125I-Tyr4-BBN being used as the radioligand. Human androgen-insensitive prostate cancer PC-3 cells are known to express GRPR in very high numbers, therefore we chose to use them for these binding assays. All of the non-metallated and metallated conjugates exhibit high or moderately high binding affinity towards the GRPR, with IC50 values in the single or double-digit nanomolar range (Table II). The binding affinities of these conjugates are, in fact, very similar to those of NOTA conjugates reported from our laboratory (32, 33).
Internalization is the accumulation of radioconjugate over time within the cells expressing the targeted receptor. The internalization behavior of each the 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 (agonist) and 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt (antagonist) conjugates were studied in human PC-3 prostate cancer cells by incubation at 37°C for 15, 30, 45, 60, 90, and 120 min and characterized by a gradually increasing rate of accumulation within the cells. The internalization rates were calculated relative to the total amount of radiotracer activity added to each well. There is a stark contrast between the agonist and the antagonist ligands in terms of the rate of internalization. For example, the percentage of cell-associated radioactivity internalized for the agonist at the 120 min time-point was 6.96±0.24. For the antagonist conjugate, the rate of internalization was 0.31±0.06% at 120 min post-incubation (Figure 2). This corroborates the fact that the agonist BBN(7-14)NH2 is endocytosed and responsible for the internalization of the conjugate. The internalization of the agonist radiotracer did not increase by extending the incubation period beyond the 90 min time point. However, for the antagonist, a gradual increase was still present at the 120 min time point. For the agonist, the percentage of internalized radioactivity exceeded that of surface-bound radioactivity at all of the investigated incubation time-points. However, for the antagonist ligand, this is not the case. For example, surface-bound tracer exceeded internalized radioactivity at all time-points. These findings are consistent with some recent studies carried out on similar type of BBN radioconjugates by our group (32, 33).
50% Inhibitory concentration (IC50) values of non-metallated and metallated conjugates in human prostate PC-3 cells (n=3).
Externalization was evaluated after an initial 45-min internalization period and was evaluated over a period of 120 min (Figure 3). The percentage of 64Cu activity remaining internalized after 120 min of incubation was 89.5% for 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 (agonist) and 52.0% for 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt (antagonist). The cell-associated radioactivity for the antagonist construct continued to decrease with each cell washing, which is indicative of the surface-bound nature of the antagonist ligand frameworks. Studies have shown that the small degree of internalized radioactivity for antagonist conjugates based upon bombesin is either a reflection of very slow internalization, or slow migration from an accessible binding region of the receptor to a more central, inaccessible binding site (11, 33). Therefore, this decrease in cell-associated radiotracer over time was expected.

Internalization rates of agonist 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 and antagonist 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt in GRPR-expressing PC-3 prostate cancer cells.
The biodistribution behavior of both the 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 (agonist) and 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt (antagonist) conjugates were initially studied in healthy CF-1 mice (Table III). For the agonist ligand, our studies were at 1 h p.i. whereas for the antagonist, the time points for evaluation were selected to be at 0.25 h and 1 h p.i. because of the limited internalization capability of the construct. The agonist conjugate showed very high uptake in normal pancreatic tissue (26.71±4.52%ID/g at 1 h p.i.), in comparison to the antagonist conjugate (4.90±0.45 and 0.27±0.07%ID/g at 0.25 h and 1 h p.i., respectively). This might suggest the effectiveness of the agonist over the antagonist conjugate for targeting of the GRPR, as it is well-known that the pancreas is the primary nonmalignant tissue expressing a high density of cell-surface GRPR in rodent models (10, 19). Minimal liver uptake was observed for the agonist (0.82±0.14%ID/g at 1 h p.i.), as well as the antagonist conjugate (2.79±0.25 and 1.07±0.08%ID/g at 0.25 h and 1 h, respectively) along with rapid clearance from blood (≤0.5%ID remaining in circulation at 1 h p.i for both of the constructs) illustrates very high in vivo kinetic stability and negligible demetallation of the Cu(II) complex. Both of the conjugates also exhibited rapid urinary excretion of the radioligand, with 75 to 88% of the radiotracer being eliminated at 1 h p.i. Each of the conjugates demonstrated favorable pharmacokinetic properties for molecular imaging and were therefore chosen to be studied in PC-3 tumor-bearing SCID mice.

Externalization rates of agonist 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 and antagonist 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt in GRPR-expressing PC-3 prostate cancer cells.
A summary of the biodistribution data of 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 and 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt in SCID mice bearing PC-3 xenografts is shown in Tables IV and V. Excretion routes were consistent for each of the conjugates and are similar to those observed for normal CF-1 mice. Each of the tracers was cleared effectively from the bloodstream, with 0.46±0.42 and 0.33±0.23%ID remaining in whole blood at 1 h p.i. for agonist and antagonist ligand, respectively. Minimal accumulation and retention of radioactivity was observed in the liver (0.60±0.18 and 0.25±0.06%ID/g) for the agonist, as well as the antagonist, constructs at 4 h p.i., demonstrating that changing the bifunctional chelator from NO2A to NODAGA did little to affect the in vivo stability of the Cu(II) metal complexes. Retention of radiotracer in normal organs for the antagonist conjugate were appreciably reduced from 0.25 h to 4 h p.i., corroborating the fact that antagonist-based constructs exhibit limited internalization. Blocking assays, which have been reported for constructs of similar structure, have indicated reduction of uptake of tracer in normal pancreas by ~90%, confirming the affinity for ligands of this type for the GRPR (34).
Rodent pancreas expresses the GRPR in very high numbers (10). The agonist conjugate exhibited significantly higher pancreatic uptake (20.42±7.18, 8.15±0.94 and 2.52±0.99%ID/g at 1, 4, and 24 h) than the antagonist ligand (6.56±1.74, 0.30±0.18, 0.04±0.03, and 0.04±0.02 %ID/g at 0.25, 1, 4, and 24 h), which could also be due to the reduced internalization of the antagonist peptide. Uptake and retention of the radiotracers in human tumors was higher for the antagonist conjugate (9.07±1.64, 5.50±1.75 and 2.08±0.23% ID/g at 0.25, 1, and 4h) as compared to the agonist ligand (4.01±1.36 and 1.21±0.27%ID/g at 1 and 4 h). Although the high uptake and retention of antagonist conjugate in the tumor tissue is not fully understood, it may well be possible that radiotracers of this general type form slowly dissociating antagonist–receptor complexes that result in longer-lasting in vivo receptor-bound radioligands. These findings are consistent with a prediction of a model for G-protein-coupled receptors which suggests that although agonist constructs have a very high affinity for the receptor sites, they only bind to a fraction of receptors associated with the G-protein, whereas antagonists can identify additional uncoupled receptors (35). Retention in normal tumor was higher for the agonist ligand at the 24 h time-point (0.80±0.12%ID/g for agonist; 0.18±0.08%ID/g for antagonist). In contrast to the moderately high uptake and retention of both the radioconjugates in the tumor, rapid clearance was observed for all other tissues. This behavior is consistent with that for some similar types of conjugates reported previously (32, 33). Some initial accumulation of the radiotracer was observed in the lung (2.49±0.11%ID/g at 0.25 h) for the antagonist construct, which decreased significantly over time. The lung accumulation could be due the normal expression of GRPR in this tissue (36). An overall negative charge for the conjugates could be the reason for relatively higher kidney uptake in comparison to similar agonist and antagonist conjugates that have been reported utilizing NOTA as a bifunctional chelating ligand (10, 32, 33).
Biodistribution of 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 and 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt in healthy CF-1 mice at 0.25 and 1 h p.i. (n=5, %ID/g ±SD).
Biodistribution of 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 in PC-3 tumor-bearing SCID mice at 1, 4, and 24 h p.i. (n=5, %ID/g ±SD).
Biodistribution of 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt in PC-3 tumor-bearing SCID mice at 0.25,1, 4, and 24 h p.i. (n=5, %ID/g ±SD).
Discussion
Diagnostically useful tumor/nontumor ratio is the prerequisite for the development of peptide based site-directed imaging agents. Moderate and diagnostically significant tumor/nontumor ratios were achieved for both of the conjugates (Table VI). For the agonist conjugate, the tumor to liver ratio range of 2.01-3.54 was comparable to those of similar 64Cu-BBN conjugates reported previously, and better than 64Cu-SarAr-SA-8-Aoc-BBN(7-14) and 64Cu-SarAr-SA-8-Aoc-GSG-BBN(7-14) that was reported recently by Lears et al. (37). This again proves the high in vivo kinetic stability of the 64Cu-NODAGA metal complex. A tumor to kidney ratio ranging from 1.0-1.56 is consistent with those for other reported 64Cu-BBN conjugates. Tumor to blood and tumor to muscle ratios were 8.71-20.0 and 23.6-120.0, respectively, which are consistent with the 64Cu-bombesin agonist conjugates reported using a variety of bifunctional chelators (10, 37, 38). Similarly, for the antagonist conjugate, the tumor to liver and tumor to kidney ratios ranged from 2.77-8.60 and 0.86-10.4, respectively, and are very similar to those for conjugates reported previously (11, 33, 34). The tumor/nontumor to blood and muscle ratios are considerably high and are comparable to 64Cu-CB-TE2A-AR antagonist-targeting vector (28). When directly compared with one another at 1 and 4 h p.i., the antagonist ligand showed superior tumor/nontumor ratios, which could be attributed to a comparatively higher tumor uptake and retention that was observed for this targeting probe. However, at the 24 h time-point, retention in normal tumor was considerably lower for the antagonist ligand framework, 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt, which resulted in lower tumor/nontumor ratios in many of the observable tissues/organs that we have reported.
Target-to-nontumor uptake ratios of 64Cu in PC-3 tumor-bearing SCID mice for 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 (agonist) and 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt (antagonist).
For many of the conjugates that we have previously investigated, diagnostically useful tumor/nontumor ratios could only be achieved at a delayed time point. Therefore, microPET/CT molecular imaging investigations using 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 and 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt were performed in PC-3 tumor-bearing SCID mice at 15 h following tail vein injections of tracer molecule. Briefly, ~600-700 μCi of each conjugate was administered to the tumor-bearing rodents. At 15 h p.i., the mice were humanly euthanized and subsequent imaging studies were performed. Molecular imaging studies show excellent microPET/CT images, with virtually no residual gastrointestinal radioactivity for the agonist construct, whereas clear visualization of xenografted PC-3 tumors was observed for the antagonist counterpart, with some degree of background and collateral radioactivity in the liver and the abdominal region (Figure 4). It is worth mentioning at this point that the chelating ligand does make a difference. Utilization of a charged ligand–metal complex, as shown in this study, could be responsible for facilitation of urinary excretion and clearance of background and collateral radioactivity from normal and surrounding tissues, which could lead to high-quality, high-contrast images. Comparing agonist to antagonist ligand frameworks in the same animal model, the agonist construct seems to be far superior in terms of microPET/CT molecular imaging. The biodistribution studies clearly reflect these findings at the later time points we evaluated. For example, compared to the agonist at 1 and 4 h p.i., the antagonist exhibits higher tumor uptake and superior tumor/nontumor ratios. On the other hand, data at 24 h p.i. suggests that the agonist ligand exhibits higher tumor retention and more superior tumor/nontumor ratios. The p-values at the 4 and 24 h time-points were less than 0.05, indicating statistically different rates of uptake/retention in tumor tissue for these two sets of data at the 95% confidence interval.
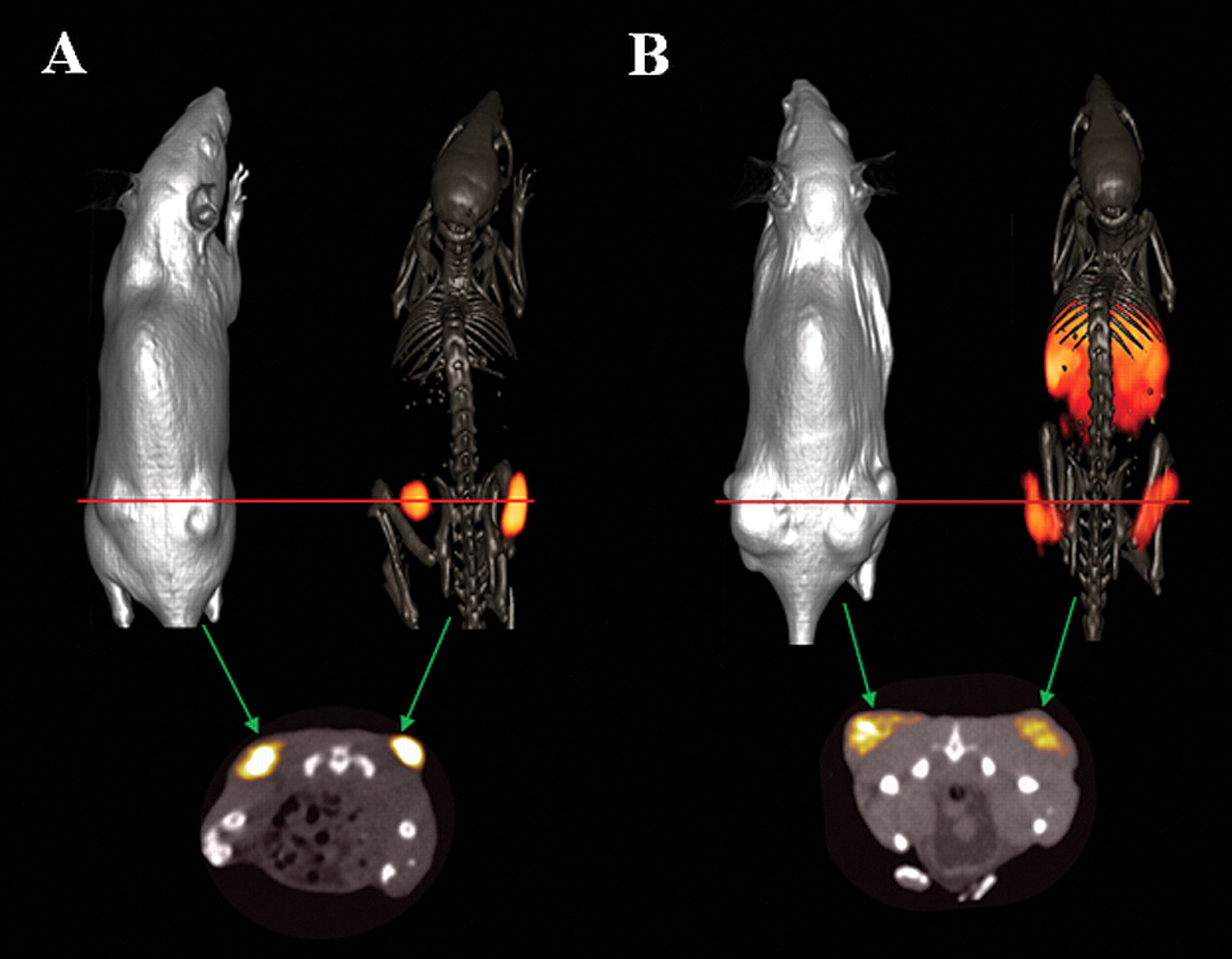
Maximum intensity whole-body microCT and microCT/PET skeletal fusion coronal images of PC-3 tumor-bearing SCID mice after 15 h tail-vein injection of 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 (A) and 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt (B).
The presence of D-Phe in the 6 position and NHEt on the C terminal position of the antagonist-targeting vector confers much hydrophobicity on the overall conjugate and could also be a reason for high background and collateral radioactivity in liver/intestine in the microPET/CT image at the later imaging time point. This could possibly be overcome by usage of a more hydrophilic pharmacokinetic modifier or placement of a NHMe group in the C-terminal position. These possibilities are currently being investigated by our research group.
In this study, we have reported the synthesis, characterization, and biological evaluation of 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2, an agonist-targeting vector having very high affinity for the GRPR. In comparison, we have also described the antagonist 64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt GRPR-targeting ligand. In these investigations, we have described in vitro assays, in vivo biodistribution, and microPET/CT molecular imaging studies. These studies were proposed in order to shed some light onto the recently growing discussions as to whether agonist or antagonist ligands are the superior GRPR-targeting radiopharmaceutical. Although biodistribution studies account for higher tumor uptake and better tumor/nontumor ratio for the antagonist ligand, the microPET/CT images clearly confirm the agonist as being a far superior molecular imaging agent for targeting the GRPR. The high-quality, high-contrast images produced using 64Cu-NODAGA-6-Ahx-BBN(7-14)NH2 agonist are comparable to those of 64Cu-CB-TE2A-AR antagonist targeting vector, which is now being considered for human clinical trials in Europe. Structural modifications of the [64Cu-NODAGA-6-Ahx-DPhe6-BBN(6-13)NHEt antagonist ligand, such as increasing the hydrophilicity, followed by an additional one-on-one detailed comparison to the agonist in the same animal model could provide a clearer perspective regarding the superiority of agonist/antagonist-based bombesin radiopharmaceuticals for molecular imaging investigations. At this point in time, however, it appears that there are many contributing factors regarding the superiority of one targeting ligand versus another for targeting GRPR-specific tissues in vivo. Clearly, the choice of targeting vector, pharmacokinetic modifier, and complexing agent for the radiometal, all play pivotal roles in uptake, retention, and excretion of the radiopharmaceutical. While one piece of the puzzle may appear to produce superior pharmacokinetic and molecular imaging data in a specific animal model, these properties may prove opposite for tracer molecules that are very similar in structure, yet altogether different in their in vivo behavior.
Acknowledgements
This material was the result of work supported with resources and the use of facilities at the Harry S. Truman Memorial Veterans' Hospital, Columbia, MO and the University of Missouri School of Medicine, Columbia, MO, USA. This work was funded in part by a United States Department of Veterans' Affairs VA Merit Award.
- Received May 30, 2012.
- Revision received June 18, 2012.
- Accepted June 19, 2012.
- Copyright © 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Development of Improved Tumor-Residualizing, GRPR-Targeted Agents: Preclinical Comparison of an Endolysosomal Trapping Approach in Agonistic and Antagonistic Constructs
- PET Using a GRPR Antagonist 68Ga-RM26 in Healthy Volunteers and Prostate Cancer Patients
- Bombesin-Targeted PET of Prostate Cancer
- N-Terminal Modifications Improve the Receptor Affinity and Pharmacokinetics of Radiolabeled Peptidic Gastrin-Releasing Peptide Receptor Antagonists: Examples of 68Ga- and 64Cu-Labeled Peptides for PET Imaging