


Abstract
Background: Multiple endocrine neoplasia (MEN) IIb is a rare genetic syndrome characterized by the occurrence of medullary thyroid carcinoma (MTC), pheochromocytoma and mucosal neuromas. Case Report: A 43-year-old woman with MEN IIb syndrome presented to our department with a painful enlargement of the left side of her vulva, which was initially presumed to be an inflammatory Bartholin's gland process. Upon admission, the patient was on antibiotics with no response and surgery was decided. A wide local excision was performed and histology revealed a metastatic medullary carcinoma of the vulva. Conclusion: MEN IIb syndrome is a clinical entity that may present multiple metastatic sites. To our knowledge, this is the first case of vulvar metastasis as part of the syndrome.
Multiple endocrine neoplasia type IIb (MEN IIb) is a rare autosomal dominant clinical entity characterized by medullary thyroid carcinoma (MTC), pheochromocytoma and mucosal neuromas (1). In 90% of patients with MEN IIb syndrome, a point mutation (ATG to ACG mutation) at codon 918 of the RET protooncogene, which codes for a tyrosine kinase receptor expressed in neural crest-derived tissues, has been observed (2). This is a rare case of a 43-year-old woman with MEN IIb syndrome who presented with a vulvar metastasis of medullary thyroid carcinoma.
Case Report
A 43-year-old woman, gravida 3, para 1, presented with recurrent left bartholin's gland inflammations during the previous nine months which were treated conservatively with antibiotics.
Her first menstrual period was at the age of 12 with a regular menstrual history. The patient's medical history goes back to the beginning of 1990, when eight weeks after an uncomplicated full-term vaginal delivery, she developed intermittent episodes of hypertension, flushing and sweating associated with anxiety, palpitations, dizziness, nausea and vomiting. Initially these symptoms were attributed to psychological depressive changes associated with the postnatal period. A few weeks later, a pheochromocytoma was suspected and confirmed by catecholamines, metanephrines and vanillyl mandelic acid (VMA) measurements. CT scanning revealed bilateral adrenal tumors. The presence of multiple neuromas of the tongue prompted the patient's physician to screen her for medullary carcinoma of the thyroid gland, as multiple MEN IIb syndrome was suspected. Plasma calcitonin measurements confirmed this hypothesis.
In 1991, the patient was operated on and bilateral adrenalectomy was performed. Two large adrenal tumors were removed and pathologically confirmed to be pheochromocytomas. One year later, the patient again underwent surgery, this time for neck exploration. A suspicious median lymph node was removed and frozen sections revealed metastatic medullary carcinoma of the thyroid. Surgery was completed with total thyroidectomy, resection of all median lymph nodes and reimplantation of two parathyroid glands found to be normal into the right sternocleidomastoid muscle.
In 1996 and 2005, multiple neuromas of the tongue and lips were excised and pathology showed no signs of malignancy. She was on medication for hypothyroidism (thyroxine 100 mg once daily) and adrenal insufficiency (hydrocortisone 20 mg once daily, fludrocortisone 100 μg once daily). Upon admission, she was on antibiotic therapy for the infection of the left bartholin's gland. Systemic examination was normal. Pelvic examination revealed normal sized uterus and adnexae. A solid fixed mass with irregular surface measuring 4-5 cm was noticed subcutaneously on the left side of the vulva, extending posteriorly into the ischiorectal fossa, which was thought to be a bartholin's gland inflammation. On investigatory hemogram, biochemical profile and chest X-ray were all within normal limits except for an elevated plasma TSH and calcitonin (9.6 μIU/ml and 49.8 pg/ml respectively).
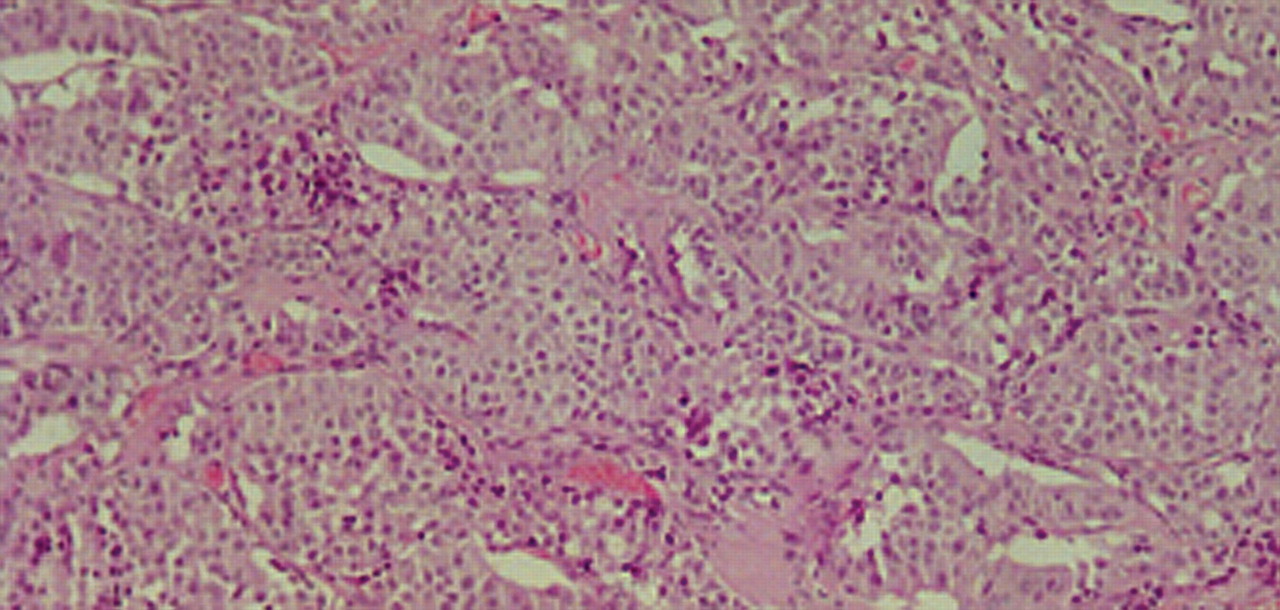
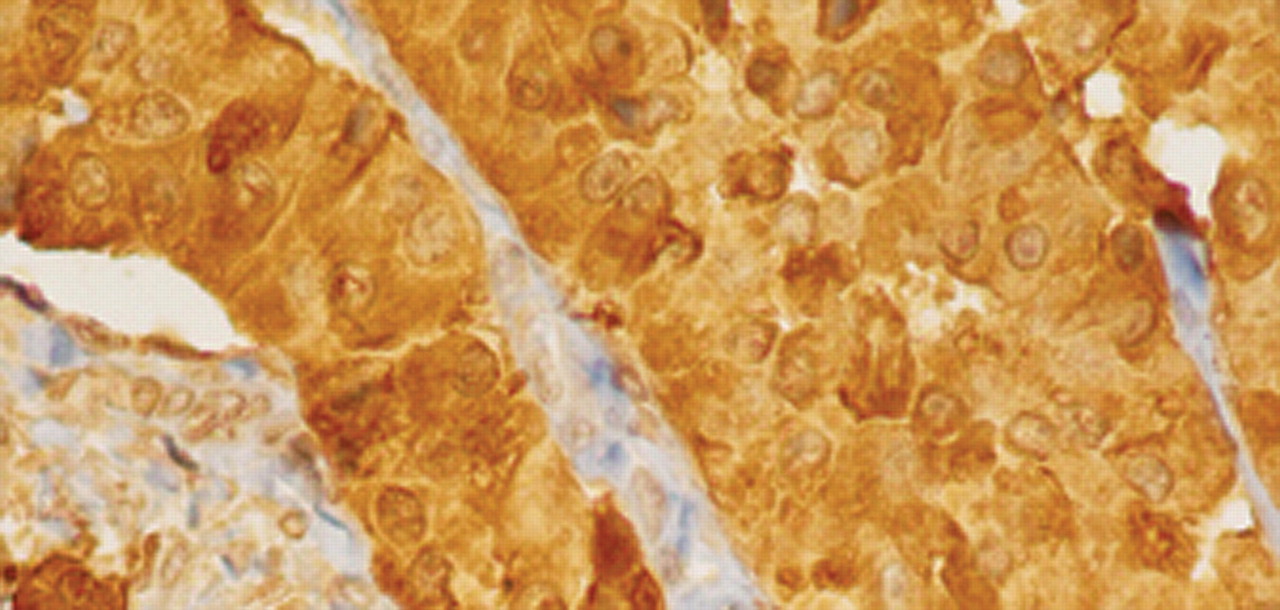
The decision was for a wide local excision of the gland. Surgery was performed and the patient made an uneventful recovery and was released from hospital after two days of hospitalization. Final histology revealed infiltration of fibrous and muscle tissue from a neoplasm with glandular pattern and oncocytic features focally. Extensive lymph vascular invasion and neurotropism were observed. Widespread amyloid deposition was identified between glandular elements (Figure 1). The neoplasm had blunt nuclear features and rare mitoses. Immunohistochemistry was positive for CK7, chromogranin, synaptophysin NSE, CEA, calcitonin (Figure 2), TTF1 and negative for ER, vimentin, GCDFP-15 and CK20. Due to the specific morphology of the neoplasm, the immunohistochemical findings and history of the patient, we have to assume that this is a case of a metastatic medullary carcinoma.
After surgery further diagnostic imaging followed, including abdominal magnetic resonance imaging (MRI), positron emission tomography/computed tomography (PET/CT) scan and iodine-131-meta-iodobenzylguanidine (MIBG). Increased uptake of the isotopes (18F-fluorodeoxyglucose and 131I-MIBG) was observed in the cervical portion of the vertebral column (Figure 4) and the anatomical position between the portal vein, the inferior vena cava and the celiac trunk (Figures 3 and 5). The latter mass had similar characteristics to an ectopic pheochromocytoma (with the differential diagnosis including a metastatic medullary carcinoma) and the patient was referred to the National Institutes of Health, Bethesda, Maryland, U.S.A. for further evaluation and treatment.
Discussion
Hyperplasia of the parafollicular C-cells is the hallmark of MEN II, with a penetrance approaching 100% and nearly all patients develop clinically apparent MTC, with a greater risk in MEN IIb syndrome (3). Concerning MTC, total thyroidectomy with central lymph node dissection should be performed (4). All patients with MTC should be evaluated for the possible presence of pheochromocytoma which should be removed first when present. MTC in patients with MEN IIb syndrome is very aggressive and surgery is often not curative (5, 6). The response to radiotherapy, chemotherapy or radioactive iodine treatment has usually been poor (4). The clinical course of patients with MTC and persistent elevation of calcitonin is very questionable. Many of these patients continue to do well, with no evidence of disease for many years after thyroidectomy and node dissection (7). Other studies report a poorer outcome in these patients (8, 9). Our patient had persistent elevation of calcitonin after thyroidectomy and node dissection and was doing well 17 years after surgery. In the absence of visible disease by imaging, patients with elevated calcitonin have two options: observation with periodic CT scanning of the chest, abdomen and neck or reoperation if they did not have an adequate central and lateral node dissection.
Pheochromocytoma occurs in approximately 40% of cases of cases of MEN IIb syndrome and rarely represents the initial manifestation of the syndrome (10, 11). In patients who have undergone regular screening, pheochromocytomas usually become evident 10 years later than MTC. Our patient, after a full-term vaginal delivery, showed typical symptoms of pheochromocytoma, such as anxiety, headache, nausea, palpitations, hypertension and tachycardia. In one report, however, only one third of patients had hypertension at the time of diagnosis (12).
Neuromas are very rarely the presenting symptom in patients with MEN IIb syndrome, although bowel obstruction caused by intestinal neuroma has been reported in the literature (13). In our case, neuromas of the tongue and lips were excised twice (1996 and 2005) and histopathology showed no evidence of malignancy. Children of patients with MEN IIb syndrome should be genetically screened for the disease. The son of our patient (20 years old) had not undergone screening. Genetic counseling in our hospital convinced her to have her son tested for the syndrome. It has been shown that prophylactic thyroidectomy based on DNA testing is very important and may provide cure for the patient (14). An important issue is the optimal time of surgery in children identified as carrying an RET mutation. According to a MEN consensus meeting in 2001, thyroidectomy and central node dissection should be performed at age 4 to 6 years (10, 15). Thyroidectomy is justified even earlier in MEN IIb syndrome, as metastatic disease in 1-year-old children has been reported (3).
Diagnostic imaging in our patient demonstrated other possible sites of metastatic disease. It would be interesting to elucidate whether these findings represent a true ectopic pheochromocytoma or a pure metastatic form of the medullary thyroid carcinoma. Only surgery and excision of the suspicious mass may provide us with the definitive diagnosis.
In conclusion, this report describes an unusual presentation of MEN IIb syndrome, many years after the initial diagnosis, and emphasizes the diversity of neoplasms that could be encountered in this disease and the complex mechanisms involved in each pathogenesis. Although apparently rare, this case highlights the need for watchful care and prompt recognition of isolated but not unexpected disorders of MEN IIb syndrome.

Medullary carcinoma: low power microscopic view with glandular and oncocytic pattern and deposition of amyloid (H-E ×100).

Intense (nuclear and cytoplasmic) calcitonin immunohistochemical stain (×400).

PET/CT demonstrating a mass measuring up to 3.2 cm in the anatomical position between the inferior vena cava, portal vein and celiac trunk.

PET/CT (cervical portion of the vertebral column).

MIBG showing the same mass as in Figure 3.
- Received April 21, 2010.
- Revision received June 16, 2010.
- Accepted June 25, 2010.
- Copyright © 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Related Articles
Cited By...
- No citing articles found.