


Abstract
Background, Materials and Methods: Synthetic triazoles are widely used for the treatment of fungal infection. In order to understand their possible anti-inflammatory action, we investigated the effect of itraconazole and its hydroxylated derivative (hydroxyitraconazole) on the production of various pro-inflammatory substances by mouse macrophage-like RAW264.7 cells. Results: These compounds did not apparently show any growth inhibitory or stimulatory effects over a wide range of concentrations (0.2-50 μg/ml). Itraconazoles dose-dependently increased the production of prostaglandin E2 (PGE2) and tumor necrosis factor-α (TNF-α) without affecting the production of interleukin-1β (IL-1β) and nitric oxide (NO). LPS treatment significantly enhanced the production of NO, PGE2, TNF-α and IL-1β. The addition of itraconazoles to LPS-stimulated RAW264.7 cells significantly reduced the production of NO, but rather enhanced the production of PGE2, TNF-α and IL-1β. ESR spectroscopy demonstrated that itraconazoles did not significantly scavenge NO and superoxide anion radicals, indicating that the inhibition of NO production by itraconazoles is not due to their radical-scavenging activity. Hydroxyitraconazole was slightly more cytostatic, and more efficiently inhibited NO production, but enhanced the production of other pro-inflammatory substances. Conclusion: These data suggest that itraconazoles regulate NO and other pro-inflammatory substances differently in activated macrophages.
The azole antifungals include two broad classes, imidazoles and triazoles, which share the same antifungal spectrum and mechanism of action. The major effect of imidazoles and triazoles is inhibition of 14-α-sterol demethylase, a microsomal cytochrome P450 (CYP) enzyme. Triazoles thus impair the biosynthesis of ergosterol for the cytoplasmic membrane and lead to the accumulation of 14-α-methylsterols. These methylsterols may disrupt the close packing of acyl chains of phospholipids, impairing the functions of certain membrane-bound enzyme systems such as ATPase and enzymes of the electron transport system and eventually inhibiting growth of the fungi (1).
Itraconazole, a synthetic triazole, is an equimolar racemic mixture of four diastereoisomers (two enantiomeric pairs), each possessing three chiral centers. The structural formula of itraconazole (Figure 1) is closely related to the imidazole ketoconazole (1). Itraconazole is widely used for the treatment of fungal infections such as skin and internal organs mycosis, and the ringworm of the nail (2, 3). Azole antifungals have been reported to affect host-mediated immune function (4). Itraconazole is metabolized in the liver and is both a substrate for and a potent inhibitor of CYP3A4. Itraconazole is present in plasma with an approximately equal concentration of a biologically active metabolite, hydroxyitraconazole (Figure 1) (1).
Hydroxyitraconazole has a similar magnitude of antifungal activity to itraconazole (5). However, no study to date has investigated the enhancement of the host defense mechanism by hydroxyitraconazole. In order to understand the role of these itraconazoles in any possible anti-inflammatory action, we investigated their effects on the production of nitric oxide (NO), prostaglandin E2 (PGE2), tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) by lipopolysaccharide (LPS)-activated mouse macrophage-like RAW264.7 cells.

Structural formula of itraconazole and hydroxyitraconazole.
Materials and Methods
Materials. The following chemicals and reagents were obtained from the indicated companies: hydroxyitraconazole, itraconazole (Janssen Pharmaceutical K.K., Tokyo, Japan), Dulbecco's modified Eagle's medium (DMEM), phenol red-free DMEM (Invitrogen Corp, Carlsbad, CA, USA), fetal bovine serum (FBS) (Gemini Bio-Products, Woodland, CA, USA), dimethyl sulfoxide (DMSO) (Wako Pure Chem. Ind., Osaka, Japan), Escherichia coli LPS (Serotype 0111:B4), 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), hypoxanthine (HX), xanthine oxidase (XOD), diethylenetriaminepenta-acetic acid (DETAPAC) (Sigma Chem. Ind., St. Louis, MO, USA); 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (carboxy-PTIO) (a spin trap agent), 1-hydroxy-2-oxo-3-(N-3-methyl-3-aminopropyl)-3-methyl-1-triazene (NOC-7) (NO gene, Kumamoto, Japan), superoxide dismutase (SOD) from bovine erythrocytes (Dojin, Kumamoto, Japan), PGE2 Express EIA Kit (Cayman Chemical Co, Ann Arebor, MI, USA), mouse IL-1 β EIA Kit, TNF-α EIA Kit (R&D Systems, Minneapolis, MN, USA).
Assay for viable cell number and NO production. RAW264.7 cells were cultured in DMEM medium supplemented with 10% FBS in a 5% CO2 atmosphere. RAW264.7 cells were cultured for 24 hours in 96-microwell plates in phenol red-free DMEM supplemented with 10% FBS containing different concentrations of itraconazole and hydroxyitraconazole. The concentration of NO in the culture supernatant was determined by the Griess method. From the dose–response curve, the concentration that reduced the NO production by 50% (IC50) by LPS-stimulated cells was determined. The related viable cell number of the attached cells was determined by MTT method. In brief, the cells were stained for 1 hour with 0.2 mg/ml MTT. The cells were dissolved in 0.1 ml DMSO and the absorbance at 540 nm of the cell lysate was determined.
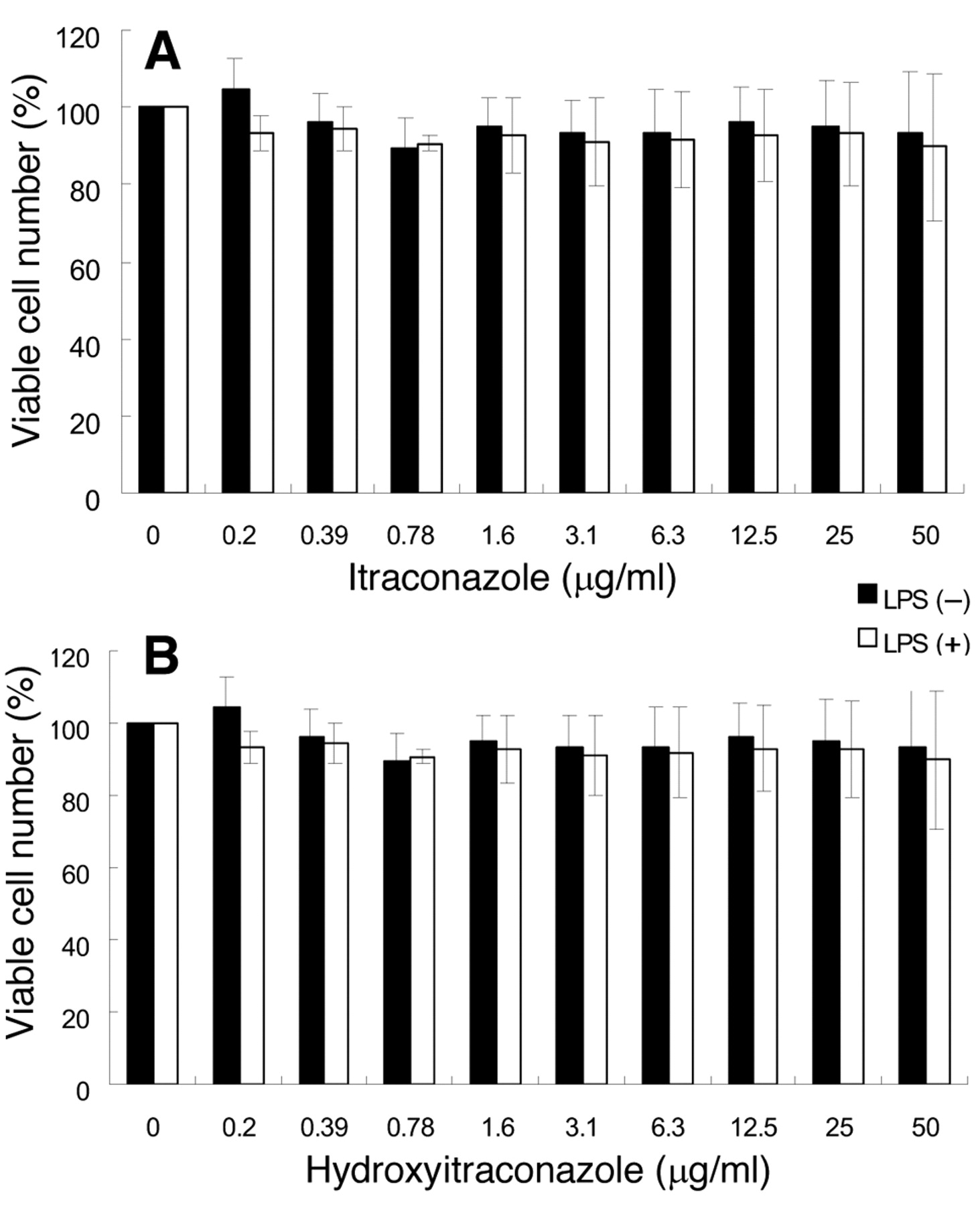
Effect of itraconazoles on the viability of unstimulated and LPS-stimulated RAW264.7 cells. RAW264.7 cells were incubated for 24 hours with the indicated concentrations of itraconazole or hydroxyitraconazole in the presence or absence of 100 ng/ml LPS, and the relative viable cell number was determined by MTT method. Each value represents the mean±SD of three independent experiments.
Assay for radical intensity. The radical intensity of the test sample was determined at 25°C, using an ESR spectrometer (JEOL JES RE1X, X-band, 100 kHz modulation frequency). Instrument settings were as follows: center field, 336.0±5.0 mT; microwave power, 8 mW; modulation amplitude, 0.1 mT; gain, 630; time constant, 0.03 seconds; scanning time, 2 minutes. The radical intensity was defined as the ratio of peak height of radicals to that of MnO. To determine O2− produced by HX-XOD reaction (total volume: 200 μl), [using 2 mM HX in 0.1 M phosphate buffer (PB) (pH 7.4) 50 μl, 0.5 mM DETAPAC 20 μl, 8% DMPO 30 μl, PB 20 μl, sample (in H2O) 50 μl and XOD (0.5 U/ml in PB) 30 μl], the gain, time constant and scanning time were changed to 500, 0.1 second and 1 minute after mixing, respectively (6, 7). For the determination of the NO radical, sample was added to the reaction mixture of 20 μM carboxy-PTIO and 50 μM NOC-7 in 0.06 M phosphate buffer, pH 7.4. The gain and scanning time were changed to 250 and 2 minutes, respectively. The NO radical intensity was defined as the ratio of peak height of the first peak of carboxy-PTI (8), produced by the reaction of NO (derived from NOC-7) with carboxy-PTIO, to that of MnO.

Effect of itraconazoles on NO production by unstimulated and LPS-stimulated RAW264.7 cells. RAW264.7 cells were incubated for 24 hours with the indicated concentrations of itraconazole or hydroxyitraconazole in the presence or absence of 100 ng/ml LPS, and the NO concentration in the medium fraction was determined. Each value represents the mean±SD from three independent experiments.
Measurement of PGE2 production. RAW264.7 cells were subcultured in 24-well plates and incubated with different concentrations of itraconazole or hydroxyitraconazole in the presence or absence of LPS (100 ng/ml). The culture supernatant was collected by centrifugation, and determined for PGE2 concentration by EIA kit.
Measurement of IL-1β and TNF-α production. RAW264.7 cells were cultured in DMEM supplemented with 10% FBS in an incubator with 5% CO2. RAW264.7 cells were cultured for 24 hours in 96-microwell plates in phenol red-free DMEM supplemented with 10% FBS containing different concentrations of itraconazole and hydroxyitraconazole in the presence or absence of LPS (100 ng/ml). The culture supernatant was collected by centrifugation, and determined for IL-1β and TNF-α concentration, according to the manufacturer's recommended procedures.
Statistical analysis. All experiments were performed in triplicate. Each value represents the mean±SD from three independent experiments (Figures 1, 2, 3, 4, 5 and 6) and from triplicate assays (Table I).
Results
Effect on growth. Itraconazole showed essentially no cytotoxicity against RAW264.7 cells over the range of 0.2-50 μg/ml (Figure 1A). Hydroxyitraconazole showed some but only marginal levels of cytotoxicity at higher concentrations. The proportion of viable cells was more than 90% (Figure 2B). Both itraconazole and hydroxyitraconazole showed little or no cytotoxicity towards LPS-stimulated RAW264.7 cells (Figure 2A, B). It has been reported that many toxicants and radiation can stimulate the growth of cultured cells at lower concentrations (so-called ‘hormesis’) (9, 10). However, we did not observe such a growth-stimulatory effect over a wide range of concentrations (Figures 2A and 2 B), suggesting a lack of hormesis in our culture system.
Radical-scavenging activity of hydroxyitraconazole and itraconazole.
Effect on NO production. Neither itraconazole nor hydroxyitraconazole enhanced NO production in RAW264.7 cells (Figures 3A and 3B). The addition of LPS (100 ng/ml) significantly enhanced NO production, up to the level of 25 μM. The addition of hydroxyitraconazole at 3.1 or 6.3 μg/ml reduced LPS-stimulated NO production by approximately 47 and 74%, respectively (Figure 3B). The IC50 was calculated to be 1.97 μg/ml. The addition of itraconazole also dose-dependently reduced NO production, but to a much lesser extent (Figure 3A). ESR analysis demonstrated that both itraconazole and hydroxylitraconasole at 1 to 1000 μg/ml, reduced the radical intensity of NO (produced from NOC-7) only by 20 and 13%, respectively (Table I). This indicates that the inhibition of NO production by itraconazoles is not due to their NO-scavenging activity.
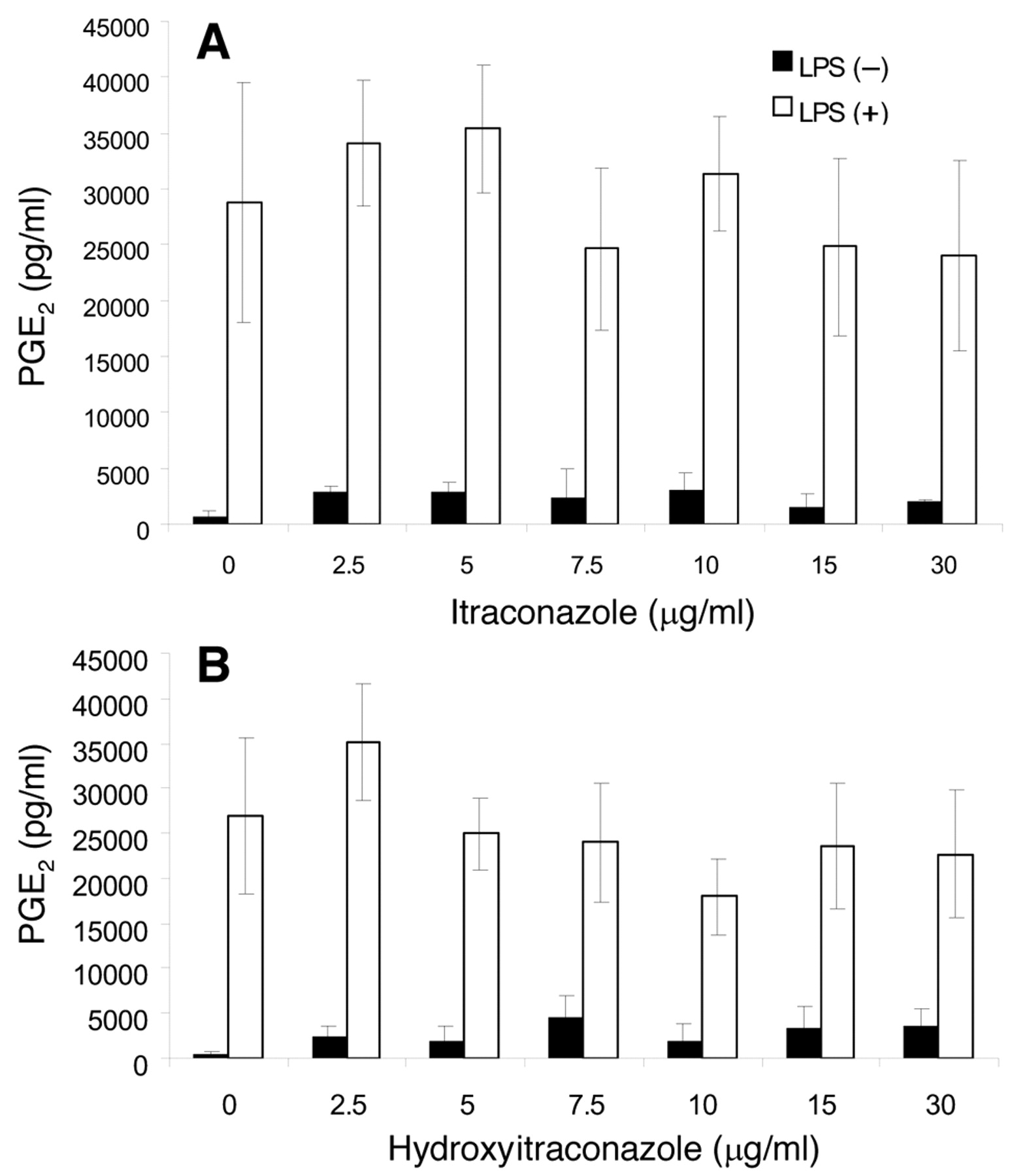
Effect on PGE2 production. Both itraconazole and hydroxyitraconazole at concentrations as low as 2.5 μg/ml stimulated PGE2 production in unstimulated cells, and this stimulation was maintained at up to 30 μg/ml (Figures 4A and 4B). The addition of LPS dramatically stimulated PGE2 production up to 30 ng/ml. Combination of itraconazoles and LPS showed some additive effects at lower concentration ranges (2.5-5 μg/ml) (Figures 4A and 4B).

Effect of itraconazole on PGE2 production by LPS-stimulated RAW264.7 cells. RAW264.7 cells were treated for 24 hours with the indicated concentrations of itraconazole or hydroxy-itraconazole in the presence or absence of 100 ng/ml LPS. The concentration of PGE2 in the medium was then determined. Each value represents the mean±SD from three independent experiments.
Effect on TNF-α production. Both itraconazole and hydroxyitraconazole stimulated production of TNF-α in unstimulated RAW264.7 cells; the stimulatory effect of hydroxyitraconazole was approximately 3 to 4 times higher than that of itraconazole (Figures 5A and 5B). The addition of LPS dramatically enhanced TNF-α production, and the further addition of both itraconazoles enhanced the LPS-stimulated TNF-α production further.
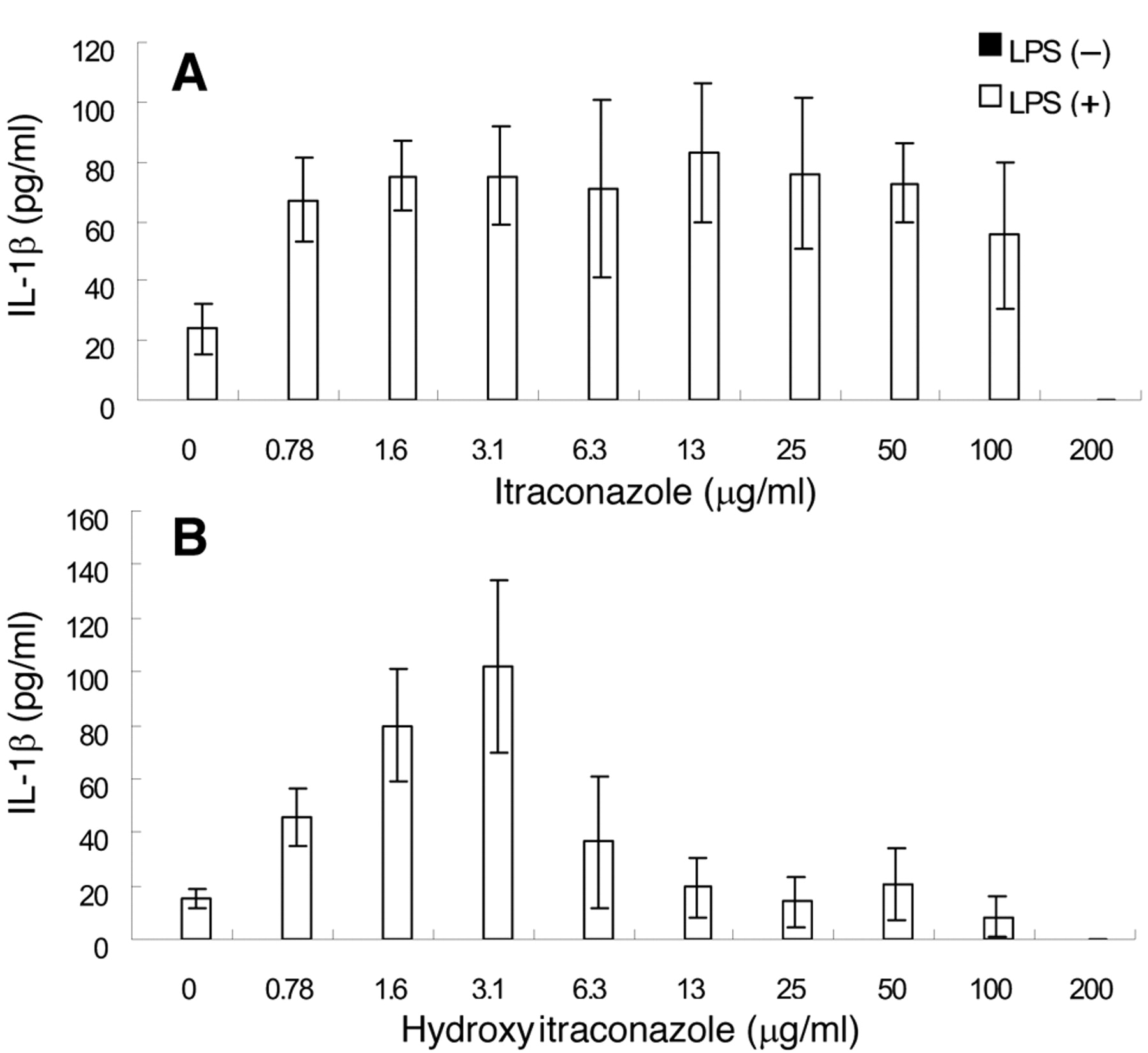
Effect on IL-1β production. Both itraconazole and hydroxyitraconazole failed to induce IL-1β production in unstimulated RAW264.7 cells, whereas LPS was found to be a potent stimulator of IL-1β production (Figures 6A and 6B). Itraconazole enhanced the LPS-stimulated IL-1β production, with the maximum effect observed at 13 μg/ml (Figure 6A). Hydroxyitraconazole stimulated IL-1β production more sharply, the optimal concentration shifting to a lower concentration (3.1 μg/ml) (Figure 6B). Our findings are summarized in Table II.

Effect of itraconazoles on the TNF-α production by LPS-stimulated RAW264.7 cells. RAW264.7 cells were treated for 24 hours with the indicated concentrations of itraconazole or hydroxyitraconazole in the presence or absence of 100 ng/ml LPS. The concentration of TNF-α in the medium was then determined. Each value represents the mean±SD from three independent experiments.

Effect of itraconazoles on the IL-1β production by LPS-stimulated RAW264.7 cells. RAW264.7 cells were treated for 24 hours with the indicated concentrations of itraconazole or hydroxy-itraconazole in the presence or absence of 100 ng/ml LPS. The concentration of itraconazoles in the medium was then determined. Each value represents the mean±SD from three independent experiments.
Regulation of the production of NO and other pro-inflammatory substance by itraconazoles.
Discussion
The present study demonstrates that both itraconazole and its hydroxy derivative when used alone stimulated the production of PGE2 and TNF-α, but did not affect the production of NO and IL-1β in RAW264.7 macrophages. An unexpected finding is that these itraconozoles enhanced the LPS-stimulated production of PGE2, TNF-α and IL-1β, but rather inhibited the LPS-stimulated production of NO. This suggests that production of NO and that of other pro-inflammatory stimulators may be differently regulated in activated macrophages (Table II). There was a possibility that the apparent inhibition of NO production by itraconazoles may simply be due to its indirect action against superoxide anion that is know to inactivate NO (11). This possibility, however, seems to be low, since we found that both itraconazoles reduced the radical intensity of superoxide anions (generated by hypoxanthine-xanthine oxidase reaction) by less than 10% (Table I).
Few comparative studies have been performed on the pharmacological actions of itraconazole and its active metabolite, hydroxyitraconazole and most of them have focused on antifungal activity (12, 13). These authors have shown comparable or higher antifungal activity of itraconazole than its hydroxyl metabolite. In contrast to previous findings, we report here for the first time that hydroxyitraconazole showed slightly higher cytotoxicity and stimulation of production of pro-inflammatory substances than did itraconazole. The reason for the discrepancy between our results and previously published findings may be due to the different culture conditions and the differences of the target cells. Further studies are necessary to elucidate the mechanism by which itraconazoles stimulate the production of pro-inflammatory substances in activated macrophages.
- Received February 26, 2010.
- Revision received July 19, 2010.
- Accepted July 28, 2010.
- Copyright © 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.