


Abstract
Background: Hepatic sinusoidal obstruction syndrome (SOS), also known as veno-occlusive disease, is a form of drug-induced liver injury, the initial morphological changes associated with which occur in liver sinusoidal endothelial cells (LSECs). Recombinant human soluble thrombomodulin (rTM) is reported to have anti-inflammatory and cytoprotective effects. Therefore, we investigated the ability of rTM to protect endothelial cells and enhance their functions in a monocrotaline (MCT)-induced model of SOS. Materials and Methods: Human umbilical vein endothelial cells were assessed in vitro following administration of MCT (2-4 mM) with/without rTM (10-100 ng/ml) to investigate the effect of rTM on cell proliferation and apoptosis. In vivo experiments were performed with Crl:CD1 mice divided into three groups: rTM (rTM + MCT), placebo (control diluent + MCT), and control (control diluent only). LSECs [cluster of differentiation (CD) 31+CD34+ vascular endothelial growth factor receptor 3 (VEGFR3)+ cells] from these mice were identified using fluorescence-activated cell sorting and assessed by quantitative real-time polymerase chain reaction (qPCR). Results: In vitro, caspase-3 and -7 activities were significantly lower and cell viability (as assessed by MTT assays) significantly higher in the rTM group than in the placebo group. Moreover, levels of p-AKT increased upon rTM administration. In vivo, damage to LSECs in zone 3 of the hepatic acinus was attenuated and the number of LSECs were maintained in the rTM group, in contrast to the placebo group. Furthermore, expression of Nos3 (encoding endothelial nitric oxide synthase) was higher and that of plasminogen activator inhibitor 1 (Pai1) lower in LSECs from mice in the rTM group than in those from the placebo group. Conclusion: rTM can attenuate SOS by protecting LSECs and enhancing their functions.
- Sinusoidal obstruction syndrome
- recombinant human soluble thrombomodulin
- hematopoietic stem cell transplantation
- liver transplantation
- oxaliplatin-based chemotherapy
Hepatic sinusoidal obstruction syndrome (SOS), also known as veno-occlusive disease, is a well-recognized and potentially life-threatening complication that occurs primarily after hematopoietic stem cell transplantation (HSCT) (1). Together with interstitial pneumonia, infections, and graft-versus-host disease, SOS has been reported to be the leading cause of death during the post-transplantation period (2), and severe SOS is associated with a mortality rate higher than 80% (2, 3). SOS has also been documented following liver transplantation, oxaliplatin-based chemotherapy, and use of pyrrolizidine alkaloid-containing herbal remedies, as well as in other circumstances, such as the autosomal recessive condition with immunodeficiency (4). Although various SOS therapies have been tested, including prophylactic anticoagulation (5), only prophylactic defibrotide treatment of pediatric HSCT patients has been shown to have an effect (6). Thus, there exist very few sufficiently effective options for SOS prophylaxis and treatment.
Pathologically, SOS is characterized by the disruption of the sinusoidal endothelium, collagen deposition in peri-sinusoidal spaces, fibrosis [especially around the centrilobular zone (zone 3) of the hepatic acinus], dilatation of the sinusoidal space, and congestion (4). The first morphological change associated with SOS occurs in liver sinusoidal endothelial cells (LSECs) of zone 3 (7); however, the events that follow are incompletely understood. We previously reported the occurrence of platelet aggregation in the extra-sinusoidal (Disse's) space and platelet phagocytosis by hepatocytes in the allograft tissue of a living-donor liver transplantation recipient with thrombocytopenia complicated by SOS (8-10). LSEC damage followed by extravasated platelet aggregation in Disse's space was a major factor in SOS progression (10-13); therefore, preservation of LSECs is of great importance in controlling the development of this disease.
Recombinant human soluble thrombomodulin (rTM) comprises the extracellular domains of thrombomodulin, and inactivates coagulation by binding to thrombin. Recently, basic research has revealed that rTM has a cytoprotective function (14), and might therefore serve as an effective treatment for SOS. In the present work, we assessed its ability to protect endothelial cells and enhance their functions in vitro and in vivo using a monocrotaline (MCT)-induced model of SOS.
Materials and Methods
Reagents. MCT (Wako, Tokyo, Japan) and rTM (Asahi Kasei Pharma, Tokyo, Japan) were used in this study.
Cell culture. Human umbilical vein endothelial cells (HUVECs) (Kurabo, Osaka, Japan) were cultured in HuMedia-EG2 (Kurabo) supplemented with 2% fetal bovine serum (FBS) (Kurabo). Protein C was not added to the culture medium.
Toxicity assay. HUVECs were seeded at a density of 5,000 cells/well in a 96-well culture plate. Twenty-three hours after seeding, rTM (10-100 ng/ml) was added to each well and cells were exposed to 2-4 mM MCT 1 h later. The concentration of MCT used was determined by referring to past literature (7). Cell viability was then assessed using a CellTiter 96 MTT assay (Promega, Madison, WI, USA), with absorbance being measured with an iMark Microplate Absorbance Reader (Bio-Rad, Hercules, CA, USA). The viability of treated cells is expressed as a fold-change compared to that of untreated control cells 8 h after MCT exposure.
Caspase activity analysis. The activities of caspase-3 and -7 were assessed with a Caspase-Glo 3/7 Assay kit (Promega, Madison, WI, USA). HUVECs were cultured in a manner similar to that used for the MTT assay, and luminescence was measured 4 h after MCT exposure using a TriStar LB 941 microplate reader (Berthold, Bad Wildbad, Germany).
Western blot analysis. HUVECs (1×106) were seeded in a 10-cm dish. Twenty-three hours after seeding, rTM (10-100 ng/ml) was added to each well and cells were exposed to 2-4 mM MCT 1 h later. The cells were lysed in radioimmunoprecipitation assay buffer containing 1% protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA) and 1% phosphatase inhibitor (Sigma-Aldrich). The concentration of protein in each lysate was measured with a bicinchoninic acid protein assay kit (Pierce Biotechnology, Rockford, IL, USA). Proteins from each sample (40 μg/well) were separated by sodium dodecyl sulfate poly-acrylamide gel electrophoresis on a 12.5% gel, before being transferred to a poly vinylidene di-fluoride membrane. The membrane was probed sequentially with antibodies against AKT (BD Biosciences, San Diego, CA, USA), p-AKT (Ser473) (BD Biosciences), plasminogen activator inhibitor 1 (PAI1) (BD Biosciences), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Animal model of SOS. Female Crl:CD1 mice (Charles River Laboratories, Yokohama, Japan) aged 6-8 weeks were used in this study. The Animal Research Committee of Kanazawa University approved all experiments (permission number 153605). The mice were divided into three treatment groups, namely, control (control diluent only), placebo (control diluent plus MCT), and rTM (rTM plus MCT), and were fasted for 12 h before drug administration. MCT (270 mg/kg) was administered by intra-peritoneal injection as previously reported (15), with some modifications. rTM (4 mg/kg) was administered by intra-peritoneal injection 1 h before MCT injection. Forty-seven hours after MCT injection, mice were sacrificed and livers were excised.
Liver histology. Liver tissues of SOS model mice were fixed in 10% neutral buffered formalin and embedded in paraffin. Slides were then made, stained with hematoxylin and eosin, and probed with an antibody against CD31 (Abcam, Cambridge, UK). Areas of CD31 staining on each slide were measured in four randomly selected images of the centrilobular zone using ImageJ (National Institutes of Health, Bethesda, MD, USA).
Liver cell isolation. Liver endothelial cells were isolated from SOS model mice with a modified two-step collagenase perfusion technique. Firstly, the portal vein was cannulated under a stereomicroscope and the inferior vena cava was cut. The liver was then perfused at 10 ml/min via the portal vein with liver perfusion medium (Invitrogen, Carlsbad, CA, USA) containing 5% FBS at 37°C for 5 min, followed by 0.05% collagenase type IV (Worthington, Freehold, NJ, USA) and 0.01% DNase (Sigma-Aldrich) for 5 min. The liver was subsequently dissociated and passed through a 100-μm nylon mesh filter. The filtrate was centrifuged at low speed (30 × g for 3 min) three times to separate hepatocytes from non-parenchymal cells (NPCs). The resulting supernatant was centrifuged three times at 300 × g for 5 min to pellet NPCs, which were then stained for cell surface markers for isolation of endothelial cells by flow cytometric analysis.
Flow cytometry and cell sorting. Isolated NPCs were incubated with Fc block (BD Biosciences) for 15 min at 4°C, before being stained with the following antibodies (for 15 min at 4°C): CD31-PE-Cy7 (BioLegend, San Diego, CA, USA), CD34-fluorescein isothiocyanate (FITC) (eBioscience, San Diego, CA, USA), and vascular endothelial growth factor receptor 3 (VEGFR3)-biotin (eBioscience). Streptavidin-allophycocyanin (APC) (eBioscience) was also added to enable detection of the latter. Cells were resuspended in 200 μl fluorescence-activated cell sorting (FACS) buffer containing 0.2 μg/ml propidium iodide (PI) (Sigma-Aldrich). LSECs (CD31+CD34+VEGFR3+PI− cells) were sorted using a FACSAria II as previously reported (16). Data were then reanalyzed with FlowJo software (Tree Star, San Carlos, CA, USA).
Reverse transcription and quantitative real-time polymerase chain reaction (qPCR). Total RNA was extracted from the sorted LSECs with an RNeasy Micro Kit (Qiagen, Germantown, MD, USA). qPCR was performed with QuantiTect SYBR Green PCR Kits (Qiagen) and an Mx3000P qPCR System (Agilent Technologies, Santa Clara, CA, USA). The sequences of the primers used for this analysis are shown in Table I.
Primers used for quantitative real-time reverse transcription polymerase chain reaction.
Statistical analysis. Differences among the data were investigated by one-way analysis of variance followed by Bonferroni multiple comparison tests or two-sided Student's t-tests with the software package IBM SPSS 24.0 (IBM Corp., Armonk, NY, USA). A p-value less than 0.05 was considered to indicate a statistically significant difference.
Results
Cytoprotective effects of rTM in an in vitro SOS model. Upon 4 mM MCT exposure, viability of HUVECs treated with 100 ng/ml rTM was significantly higher compared to that of the non-rTM treated group. However, this phenomenon was not observed with 2 mM MCT exposure. There was also no significant difference between non-rTM and 10 ng/ml rTM groups exposed to 2 mM or 4 mM MCT (Figure 1A). On the contrary, on treatment with 4 mM MCT and 100 ng/ml rTM, caspase-3 and-7 activities were significantly lower compared to those in the group without rTM treatment. However, there was no significant difference between the group without rTM treatment and groups treated with 10 ng/ml rTM and 2 mM or 4 mM MCT (Figure 1B). In addition, western blotting revealed that the p-AKT level increased in the 100 ng/ml rTM-treated group. Furthermore, level of PAI1, which represents endothelial cell damage, was reduced in the rTM-treated group compared to those in the group without rTM treatment (Figure 1C).
Macroscopic and microscopic examination of mouse livers and evaluation of endothelial damage. Macroscopically, a pale, patchy, and spotted liver surface was observed in the placebo group, but not in the rTM or control group (Figure 2A). Furthermore, liver perfusion via the portal vein revealed obvious effects on congestion (Figure 2B). Microscopically, MCT-induced liver congestion and sinusoidal dilatation were seen to be remarkably attenuated by rTM treatment (Figure 2C). ImageJ analysis showed that areas occupied by CD31+ endothelial cells in zone 3 were clearly smaller in the placebo group (Figure 2D-F). MCT treatment resulted in fewer CD31+PI− (i.e. live endothelial) cells; however, this effect was negated by administration of rTM, as assessed by FACS (Figure 2G).
Evaluation of sorted mouse LSECs by qPCR. Fatty acid binding protein 1 (Fabp1) and cytokeratin 19 (Ck19) were expressed at low levels in sorted LSECs compared to whole liver tissue, indicating that the sorted cells were indeed distinct from hepatocytes and biliary epithelial cells (Figure 3A and B). Increased expression of Pai1, Vegfa, and angiopoietin 2 (Angpt2) induced by MCT in sorted LSECs was significantly inhibited by rTM treatment (Figure 3C-E). Moreover, expression of Nos3 [encoding endothelial nitric oxide synthase (eNOS)] was reduced by MCT exposure, but maintained in cells from mice administered both MCT and rTM (Figure 3F). Hepatocyte growth factor (Hgf) expression differed little between each group, whereas Wnt2 was down-regulated in both the placebo and rTM-treated groups (Figure 3G and H).

A: An MTT assay was performed using human umbilical vein endothelial cells (HUVECs) seeded at a density of 5,000 cells/well in a 96-well culture plate. Twenty-three hours after seeding, recombinant human soluble thrombomodulin (rTM) was added to the wells, followed by 2-4 mM monocrotaline (MCT) 1 h later. Treatment with 100 ng/ml rTM significantly preserved the viability of HUVECs exposed to 4 mM MCT (n=7). Significantly different at *p<0.05. B: Caspase-3 and -7 activity was measured in HUVECs seeded at a density of 5,000 cells/well in a 96-well culture plate. rTM and MCT were administered as MTT assay. Treatment with 100 ng/ml rTM significantly reduced the elevated caspase activity caused by exposure to 4 mM MCT (n=3, *p<0.05). C: Levels of p-AKT were increased and those of plasminogen activator inhibitor 1 (PAI1) (marker of endothelial damage) reduced by administration of 100 ng/ml rTM. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control (40 μg protein/well).
Discussion
SOS is a clinically important complication given its close relationship with the morbidity and mortality of diseases necessitating HSCT, liver transplantation, or oxaliplatin-based chemotherapy, with which it is associated. Various factors are involved in the pathogenesis of SOS, including LSEC glutathione depletion, nitric oxide depletion, and increased matrix metalloproteinase (MMP) and VEGF levels (17). In our prior study, endothelial damage and platelet aggregation in the extra-sinusoidal (Disse's) space were shown to be highly important in exacerbation of SOS (10-13). Here, we focused on the protection of LSECs from toxic agents, and the cytoprotective effect of rTM in particular.
According to previous research, alteration of LSEC morphology and degradation of the sinusoidal lining disturb hepatic microcirculation (18). Clinical trials of heparin and prostaglandin E1 as treatments for the prevention of SOS based on enhancement of hepatic microcirculation have been conducted. However, a meta-analysis of 12 studies using low-molecular-weight or unfractionated heparin as SOS prophylactics showed that anticoagulation does not significantly reduce the risk of this disease (5), and a phase I/II study of prophylactic use of prostaglandin E1 was complicated by excessive toxicity and failed to show any benefit of its administration (19). Therefore, treating hepatic microcirculation dysfunction caused by LSEC injury is insufficient to prevent SOS.
Recently, active protection of LSECs has come to be considered a more effective approach to SOS treatment, especially as a preventive measure. For instance, Miyata et al. reported that a phosphodiesterase III inhibitor attenuated SOS in a rat model of this condition due to its cytoprotective effects (13). In a retrospective clinical study by Ribero et al. it was demonstrated that addition of bevacizumab, a recombinant human monoclonal antibody against VEGFA, to chemotherapy with 5-fluorouracil plus oxaliplatin led to a reduction in the incidence and severity of oxaliplatin-induced SOS (20). Notably, bevacizumab is thought to attenuate damage to LSECs by down-regulating MMP production. Furthermore, a prospective phase III study showed reduced incidence of SOS among pediatric HSCT patients having received prophylactic defibrotide (21). Defibrotide is a single-stranded polydeoxyribonucleotide whose effects are brought about by anti-thrombotic and anti-inflammatory activity and protection against endothelial injury (1). Therefore, protection against LSEC damage and preservation of endothelial cell function will be a useful SOS treatment strategy, and rTM is expected to become a new preventive and therapeutic option for this condition.

A: Female Crl:CD1 mice were divided into three groups: Control, Placebo [270 mg/kg monocrotaline (MCT)], and recombinant human soluble thrombomodulin (rTM) (270 mg/kg MCT plus 4 mg/kg rTM). rTM was administered 1 h before MCT exposure. After 48 h, macroscopic observation of livers in the placebo-treated group revealed congestion and remarkable ascites. B, C: Following perfusion of the liver via the portal vein with liver perfusion medium, congestion in the placebo-treated group was particularly notable. D: Immunostaining showed clear decrease in the area occupied by cluster of differentiation (CD) 31+ endothelial cells in zone 3 in the placebo-treated group. E, F: Areas of CD31 staining in four randomly selected images of the centrilobular zone per sample were measured using ImageJ (n=4). G: Numbers of viable CD31+ cells were established by fluorescence-activated cell sorting. The number of CD31+ propidium iodide (PI)− cells decreased due to MCT administration, but was maintained by rTM treatment (n=6). Significantly different at *p<0.05, and **p<0.01.
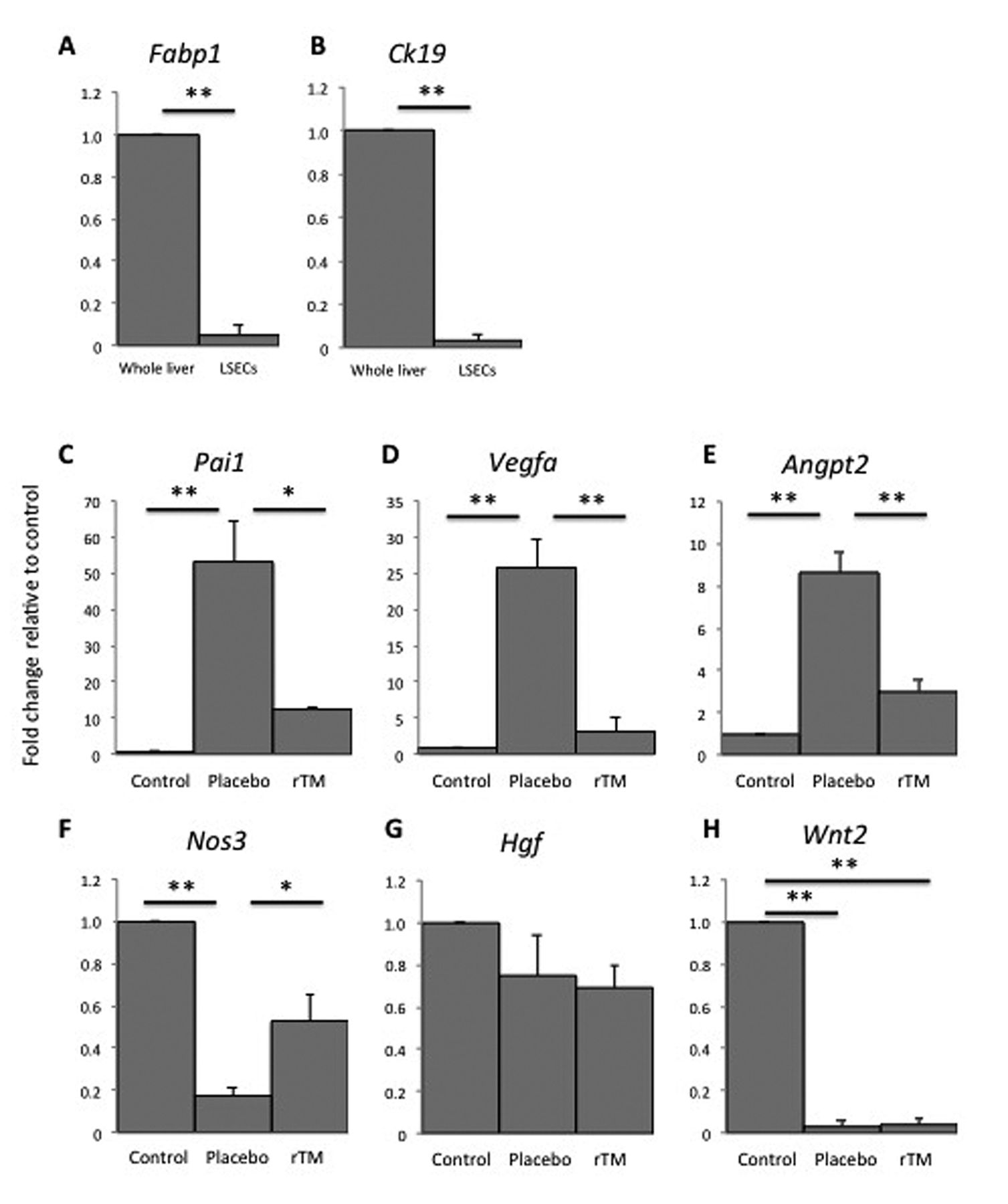
Expression of genes relative to β-actin. A, B: Fatty acid binding protein 1 (Fabp1) and cytokeratin 19 (Ck19) expression was very low in sorted liver sinusoidal endothelial cells (LSECs) compared to whole liver tissue. C-E: Expression of the genes plasminogen activator inhibitor (Pai1), vascular endothelial growth factor A (Vegfa), and angiopoietin 2 (Angpt2) was increased in the placebo [monocrotaline (MCT)] group, but significantly inhibited in the recombinant human soluble thrombomodulin (rTM) (MCT+rTM) group. F: Expression of the nitric oxide synthase 3 (Nos3) gene was preserved in the rTM-treated group. G: Hepatocyte growth factor (Hgf) expression was relatively unaffected by the different treatments. H: Wnt2 expression decreased in both the placebo- and rTM-treated group (n=3). Significantly different at *p<0.05, and **p<0.01.
rTM was approved as a standard treatment for disseminated intravascular coagulation in Japan in 2008. rTM consists of the extracellular domains of thrombomodulin, which include the lectin-like domain (D1), epidermal growth factor (EGF)-like domain (D2), and Ser/Thr-rich domain (D3) (14). The lectin-like domain binds to thrombin and inhibits binding of high mobility group box 1 (HMGB1) and lipopolysaccharide to cells and neutrophil adhesion to endothelium. The EGF-like domain generates activated protein C, protects vascular endothelial cells, and inhibits PAI1, complement factors, and coagulation. Thus, rTM possesses many important anti-coagulant and anti-inflammatory attributes (22).
From a clinical perspective, there have been several case reports of favorable outcomes from rTM treatment of patients with post-HSCT SOS complicated by disseminated intravascular coagulation (23, 24). Moreover, Nakamura et al. using an MCT-induced murine model indicated that rTM attenuates SOS through suppression of HMGB1 (25). Ikezoe et al. also reported that rTM exerted a cytoprotective effect against cyclosporine- and FK506-induced endothelial cell damage (14). Their in vitro assay revealed that rTM increased the level of the anti-apoptotic protein myeloid cell leukemia 1 via extracellular signal-regulated (ERK) signaling to protect HUVECs. Besides ERK signaling, rTM has also been reported to activate the phosphatidylinositol 3-kinase/AKT survival pathway and suppress the c-Jun N-terminal kinase death pathway (26). In the current work, we focused on AKT signaling owing to its broad effects on cell proliferation, survival, and functions (27). Furthermore, inhibition of apoptosis and activation of cell-survival signaling play important roles in the maintenance of endothelial cell viability (28, 29). Using our in vitro system, we found that the cytoprotective effects of rTM may be mediated by inhibition of caspase activity and activation of AKT signaling, both leading to anti-apoptotic effects in HUVECs. MCT toxicity was found to be concentration-dependent and a protective effect was evident upon 100 ng/ml rTM administration. Upon 2 mM MCT exposure, HUVEC viability was not significantly affected because of limited damage; however, upon 4 mM MCT exposure, significant endothelial cell preservation was observed. These results might support the utility of rTM under conditions of severe SOS.
In this study, MCT-administered mice were utilized as an experimental model of SOS (15). Liver congestion and sinusoidal dilatation induced by MCT were remarkably attenuated by preventive treatment with rTM. In addition, liver perfusion via the portal vein led to notable findings concerning congestion, and liver damage was associated with reduced areas of CD31+ endothelial cells. For the first time, we employed a multi-color flow cytometric sorting method to isolate MCT-injured mouse LSECs and analyzed their expression of a set of genes by qPCR. LSECs were sorted as CD31+CD34+VEGFR3+ PI− cells, as in a past study (16). LSEC expression of the genes Pai1, Vegfa, and Angpt2, the products of which are markers of endothelial cell damage, was increased by MCT administration, and this effect was significantly inhibited by rTM treatment. PAI1 is also used as diagnostic marker and predictor of the severity of SOS (1); thus, its down-regulation may be highly beneficial for SOS treatment. In addition, expression of the gene encoding eNOS following MCT treatment was preserved by rTM administration, indicating maintenance of LSEC viability and function in the injured liver. DeLeve et al. reported that reduced hepatic NO production contributes to the development of SOS (30). Using a rat model, they demonstrated that infusion of an NO donor preserved LSEC integrity and prevented SOS. They concluded that the decrease in NO allowed up-regulation of MMP activity and subsequent disruption of sinusoidal perfusion. Fulton et al. identified AKT as an important regulator of eNOS (31). In our study, rTM induced up-regulation of p-AKT and the gene encoding eNOS, and exhibited the ability to modulate endothelial functions that may be beneficial in SOS therapy. Given their importance in liver regeneration, we also investigated expression of the genes Wnt2 and Hgf in LSECs; however, no significant differences were noted between the placebo and rTM groups. As our mouse model represents the acute phase of SOS, a chronic-phase model might be more appropriate for assessing changes in Wnt2 and Hgf expression.
In conclusion, rTM has a cytoprotective effect and enhances endothelial cell functions, and may thereby prevent the development of SOS.
Footnotes
This article is freely accessible online.
Conflicts of Interest
The Authors declare they have no financial or other conflicts of interest in relation to the content of this article.
- Received August 11, 2018.
- Revision received September 22, 2018.
- Accepted September 25, 2018.
- Copyright© 2018, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved




{kind=link}
{kind=link}
{kind=link}