


Abstract
From 2005 to 2010, eight families with clustering of Hodgkin's lymphoma and other lymphoproliferative disorders were found: Hodgkin's lymphoma 9 cases, chronic lymphocytic leukemia 8, non-Hodgkin's lymphoma 3, and multiple myeloma 1 case. Seven cases of Hodgkin's lymphoma, all males, were seen in pleiotropic pairs of affected family members from two successive generations; two patients were sisters. Five of the seven pairs showed sign of anticipation. The 7 males with Hodgkin's lymphoma were found in 5 patrilineal pairs and 2 matrilineal pairs; 6 parent–offspring pairs and 1 uncle–nephew pair. In contrast to the matrilineal pairs, all patrilineal pairs, apart from one family with an only child, had healthy older siblings in accordance with a birth-order effect. The association among Hodgkin's lymphoma, males, and other lymphoproliferative disorders undoubtedly reflects genotypic traits of the susceptibility. A non-Mendelian segregation is discussed comprising genomic parental imprinting and incomplete penetrance susceptibility in both familial and solitary cases.
- Hodgkin's lymphoma
- chronic lymphocytic leukemia
- genetic segregation
- pleiotropy
- genomic imprinting
- birth-order effect
- anticipation
Familial clustering of lymphoproliferative disorders (LPD) has been fully confirmed since 1947, when Videbaek described two brothers, one with chronic lymphocytic leukemia (CLL) at the age of 45, and the other with Hodgkin's lymphoma (HL) at the age of 29 (1). LPD comprises malignant monoclonal lymphoproliferative disease of all kinds, such as acute lymphoblastic leukemia (ALL), CLL including chronic lymphocytosis of uncertain significance, non-Hodgkin's lymphoma (NHL) and Hodgkin's lymphoma (HL), multiple myeloma, and progressive monoclonal gammopathy of uncertain significance. Interpretation of bulk-data from cancer registries with systematic crosscheck of first-degree relatives (2-7), and analyses of pedigrees from LPD-affected families in case histories and case–control studies, both from older reports (8, 9) and from recent published cases (10-13), make it relevant now to regard LPD as a genetic entity, in which the same genes encode different manifestations, so-called pleiotropy. Different manifestations in familial coexpression of LPD have indeed been disclosed. In a large survey, the relative risk (RR) for HL among first-degree relatives of LPD patients is 3.7 for men and 1.0 for women, with a remarkably different RR for CLL in first-degree relatives (men 6.9, women 8.6), for NHL (men 1.4, women 1.5) and for multiple myeloma (men 2.8, women 0.5) (2), but without significantly increased risk for NHL among siblings of patients with HL (4). In siblings of patients with HL, the RR for HL is more than five-fold increased (6), while the risk of HL in monozygotic twins is about 100-fold increased and higher than in dizygotic twins (14), which also strongly supports a genetic etiology for HL.
Unlike sporadic cases of HL, familial HL is generally seen in younger patients, but with the same frequency of the histopathological subtypes as seen in non-familial, sporadic cases (10), and with the same response to treatment, including the same rate of cure as in non-familial HL (15). In such families, anticipation confers an increasingly lower age at onset of disease and an increased severity of disease down through the generations. In other words, a sort of genetic enhancement which has been ascribed to amplification of abnormal tri- and poly-nucleotide repeats inside the susceptibility genes (16-18). Anticipation has been seen when comparing the age at onset of LPD in large series of parent–offspring pairs and grand parent–parent–offspring combinations with HL (19-21), while others have failed to find anticipation in familial HL (16). For comparison, anticipation has also been discussed for CLL, where it is hardly present at a statistically significant level when CLL is leading, viz. parental, or the only diagnosis of the pair (22).
Concomitant lymphoproliferative disease in familial Hodgkin's lymphoma.
Candidate susceptibility loci have been described for both HL (23, 24) and CLL (25, 26) and most likely, the united LPD susceptibility genome is a polyallelic pool of genes available for meiosis under constant modulation from anticipation and under epigenetic influence, giving rise to a birth-order effect with a male predominance (27). Apparently, the HL genome has a weak association with HLA, both class I, and class II, especially DRB1, DQA1 and DQB1 loci (28-30). Theoretically, this modulating pool of susceptible polygenes has a huge capacity to segregate into a large number of hybrid-mosaics, one or perhaps a few hierarchical repeats specific for each of the different LPD diagnoses with a varying power of penetration. This genotypic pool of susceptibility represents the predisposition to mutation(s) in the growth-and-differentiation genes of the lymphoid stem cell (31). From this initial lesion, an autonomic lymphocytic monoclone may emerge and develop into manifest LPD, depending on e.g. age, gender and autoantigeneic drive, and sometimes on epigenetic environmental hits such as stimulation from lymphotrope virus such as Epstein-Barr virus (EBV), and stimulation from the antigeneic drive of many other types of infections (31-33). In a discussion of different putative genetic and infectious etiologies in HL and NHL, mutations in mismatch repair genes have been pointed out as one likely germline alteration involved in LPD clustering, which also provide an explanation for the early-onset CNS tumors in such families (6). In other studies, germline FAS mutations have been related to the increased rate of HL and NHL in familial autoimmune disease (34). Due to the inflammatory component of HL, it is difficult to ascribe a certain genetic defect either to the real, inborn susceptibility-related genotype, or, alternatively, to genes coding for the phenotypic inflammatory and reactive manifestations during tumorigenesis (32, 35, 36).
However, from a genealogical point of view, the segregation of the LPD-susceptibility genes is largely unknown, clearly non-Mendelian as already stated in 1983 (8), and most likely under epigenetic influence (8, 22, 27). The purpose of the present paper is to discuss the segregation of HL from a genealogical point of view and to outline some main characteristics.
Patients and Methods
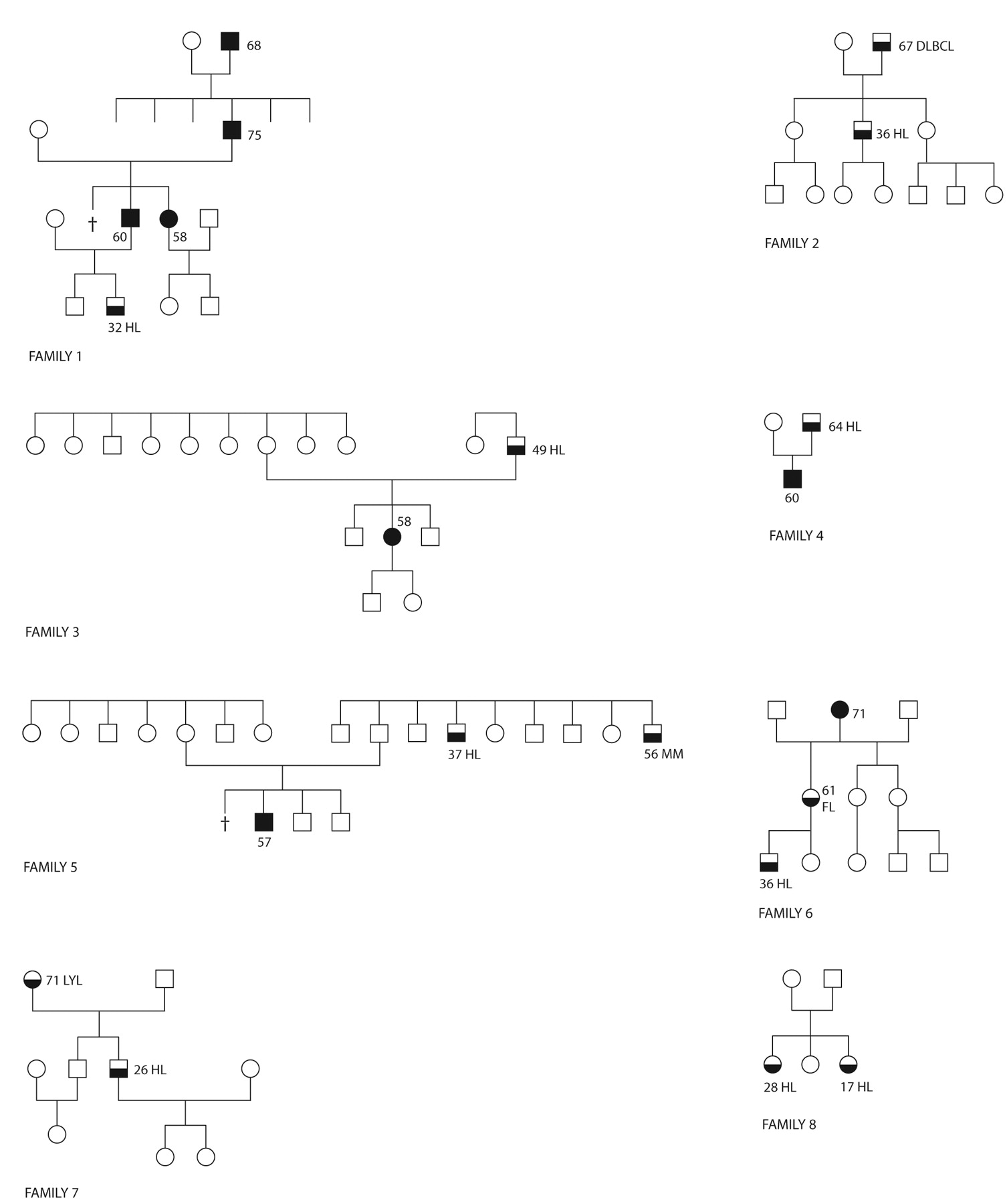
Families with Hodgkin's lymphoma. Our database on familial LPD has eight families with HL from January 2005 to June 2010. Five families (Figure 1, no. 3, 4, 5, 7 and 8) are of Norwegian origin, two families (no. 1 and 2) are Danish ,where the father with DLBCL in family no. 2 is living in Norway, and one family (no. 6) is Swedish, with the proband living in Norway. Seven families have affected family members in two or more generations (Figure 1, Table I) and one family presents a sibling concordance: Two sisters, born 1977 and 1988, both diagnosed with HL the same day in January 2005. The eight HL families have been extracted from a total of 65 families with two or more cases of LPD from the same period.
To ensure maximal ascertainment of familial cases, each LPD patient underwent an interview about other family members with HL or any other malignant hematological disease and the family tree was drawn. Each patient was asked about the number and position of healthy family members, stillborns and extra marital individuals. All patients were allowed sufficient time for discussion with other family members. Both patients and healthy family members were informed about the purpose of the study and that data were confidential and unrecognisable outside of the study. The study was approved by the Regional Committees for Medical Research Ethics in Norway.

Eight families with Hodgkin's lymphoma and other lymphoproliferative disease. Signature: The figures within the pedigrees indicate the age at onset of disease. Black, CLL; white, healthy; white to black from top to bottom indicates lymphoproliferative disorder other than Hodgkin's lymphoma. CLL, Chronic lymphocytic leukemia; DLBCL, diffuse large B-cell lymphma; FL, follicular lymphoma; HL, Hodgkin's lymphoma; LYL, lymphoplasmocytic lymphoma; MM, multiple myeloma; †death as a child.
Information provided by patients was cross checked with the Cancer Registry in Norway and with the hospital records. Histopathological and laboratory reports, including information from flow cytometry and cytogenetics, were reviewed when available. The healthy family members were cross checked and confirmed by the Civil Person Registry of Norway to ensure the right position of each person in the pedigree, including possible extra-marital children.
About 25 persons were examined per family together with the proband's parents, siblings and children, mainly maternal and paternal uncles and aunts, cousins and their children etc., giving a total of about 200 persons screened within the eight HL-families.
Concomitant LPD. The LPD diagnoses of the eight families (Figure 1, Table I) are based on standard criteria (37). There were 12 cases of LPD other than HL: 8 of CLL, 3 of nonHodgkin's lymphoma, and 1 of multiple myeloma.
Results
A total of 9 cases of familial LPD with HL were found in 8 families (Figure 1, Table I), corresponding to 8 (12%) HL-affected families from a cohort of 65 families with clustering of LPD. There were 7 male and 2 female HL patients in the 65 families, which is significantly more than expected when comparing with the occurrence of HL in the general population. Calculations based on the age-adjusted incidences (2.8 male HL patients per 100,000 persons per year and 1.4 female HL patients per 100,000 persons per year in Norway, 2008 (38) produce an expected number of HL in the 65 families with 1,625 persons (25 cross-checked persons per family) which is significantly lower (p<0.0001) with any relevant statistical model applied: 45.5×10−3 male HL patients and 22.8×10−3 female HL patients per year, compared with the 1.6 males and 0.4 females per year (7 males and 2 females during 4.5 years) seen in the families.
Seven HL cases, all males, were seen in pleiotropic pairs of affected family members from two successive generations, and two HL cases were sisters. The 7 males with HL were found in 5 patrilineal pairs and 2 matrilineal pairs, 6 parent–offspring pairs and 1 uncle–nephew pair. The male predominance in familial HL observed in the present investigation is in accordance with the male predominance seen in the age-adjusted incidences of HL. The male predominance is further reinforced by the predominant patrilinieal occurrence of HL in the transmission of LPD: 5 patrilineal pairs (Figure 1, no. 1, 2, 3, 4, 5) in contrast to only 2 matrilineal pairs (Figure 1, no. 6 and 7).
The 9 HL patients were seen among 12 family members with LPD other than HL where CLL was predominant (8 cases of CLL) and CLL was one of the two diagnoses in 4 (Figure 1, no. 1, 3, 4 and 5) out of 7 pairs with HL. In spite of the small numbers and only 8 families investigated, 8 cases of CLL (5 males and 3 females) among the 200 persons screened from the 8 families is a very high occurrence when compared with the age-adjusted incidence of CLL in Norway [7.3 male CLL patients per 100,000 persons per year and 3.2 female CLL patients per 100,000 persons per year (38)] linking familial CLL and HL closely. Based on data from these incidences, the expected numbers of CLL patients among 200 persons per year is significantly lower (14.6×10−3 males and 6.4×10−3 females, p<0.001).
Discussion
Other reported families with HL have shown a predominant patrilineal transmission, e.g. two affected boys and an affected father (11), and three affected boys with the same unaffected father but different mothers (13). A similar stronger familial association among men than among women is also seen in NHL (2, 4, 39) and in CLL (27), but not in familial monoclonal gammopathy of uncertain significance and myeloma (40). This association among HL, men, and LPD in both HL-LPD and LPD-HL pairs is in favor of a shared genotypic susceptibility. Since no data yet relate LPD susceptibility to the Y chromosome, a non-Mendelian segregation with genomic, parental imprinting seems likely (8, 41-43).
Genomic imprinting is a parental-specific gene expression based on intrauterine regulation of the fetal genetic material, giving rise to monoallelic genes depending on the paternal or maternal origin of the allele. Imprinted genes are monoallelically expressed and regulated independently of spermiogenesis and oogenesis by allele-specific epigenetic modifiers (silencer), where DNA methylation and modifications of histones are well described mechanisms (44-46). Genomic imprinting enables the female to transfer selected alleles to her offspring in a birth order. Such a birth-order effect has recently been discussed in relation to CLL and LPD, where patrilineal CLL is mainly given to the youngest in the sibship while matrilineal CLL is randomly distributed in the sibship (27). A birth order is also seen in the present material in an excess of males where the HL-affected offspring in all patrilineal combinations, except for the family with an only child, have a healthy older sibling. This mechanism supposedly secures the transmission of susceptibility genes to the offspring under the condition that paternal genes are suppressed and restricted to males and to the youngest children, supported by anticipation, which also tends to make the susceptibility genes available for the youngest. In both the matrilineal pairs, and in 3 out of 5 patrilineal pairs of the present material (Table I), anticipation is clearly seen with regard to the lowest age at onset of disease in the offspring, while anticipation with regard to increased aggressiveness of disease down through the generations is hardly visible since no high-grade malignant LPD diagnosis, for example ALL, were found.
‘Outbreak’ of HL among siblings has been related to EBV infection (7, 12, 47-50). The two sisters with HL reported in family no. 8 (Figure 1) are EBV negative, viz. in situ hybridization for EBV in the lymphoma tissue was negative. To our knowledge, these cases are unusual with regard to the female concordance and the simultaneous onset of disease. Most reported siblings with HL are males (11-13) or brother-sister combinations (10, 12), with or without in situ EBV expression by the tumor cells, two cases being EBV negative, namely the present family no. 8, and a familial case of Indo-Iranian origin (10). We find it hard to believe that EBV or any other type of environmental stimulation can exert its action alone and without an inborn susceptibility and we presume that the etiology of solitary cases of HL and the rare cases of post-transplant HL (51, 52) are also dependent on congenital susceptibility, having a low or incomplete penetrance in unaffected and healthy family members so that seemingly, no other member of the family has LPD (16, 31). Using computed likelihood analysis of the descent structure in HL pedigrees, Thompson estimated as early as 1981 that the genotype in HL has a recessive nature (53). The existence of carriers, e.g. unaffected parents between an LPD-affected grandparent and an LPD-affected child, fits in with the concept of low-penetration susceptibility in healthy family members. Such carriers have been pointed out in pedigrees with familial CLL and NHL (16, 27). In other words, solitary cases of HL and other types of solitary LPD seem to be identical with familial cases, with the exception that the power of penetration of the susceptibility genes in families with solitary cases is generally low. No differences can be seen in the cytogenetic profile of solitary and familial CLL (25), nor has any difference in the outcome of treatment between solitary and familial LPD been recorded (15). This supports the idea that solitary and familial LPD of all kinds are united in the same entity. Regarding HL, no genealogical investigation of cross checked LPD in major consecutive cohorts of HL has been published so far. To rely on such a survey, it must be based on a large LPD cohort and also include low-grade subsets of LPD with no or nearly no symptoms, e.g. chronic lymphocytosis of uncertain significance, monoclonal gammopathy of uncertain significance, stage A CLL and stage IA HL, to ensure that cases of LPD with low penetrance are also detected and included in the calculations.
Acknowledgements
We are grateful to the patients and their families for their participation in this study. Signe Nøsterud and Birgit Skjelvik are thanked for excellent technical assistance. The study was undertaken in accordance with the Declaration of Helsinki. We thank the Norwegian Data Inspectorate (record no. 07/00254-2), the Social and Health Directorate in Oslo (record no. 07/324), and the Regional Committees for Medical Research Ethics in Norway (record S 06353b) for permission to carry out the study and for access to data. We are grateful to the Institute of Clinical Medicine, University of Oslo, for financial support.
- Received November 30, 2010.
- Revision received January 31, 2011.
- Accepted February 1, 2011.
- Copyright © 2011 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.