


Abstract
Background: Multidrug resistance (MDR) is one of the major concerns in the treatment of cancer and one of the major causes of therapy failure. The overexpression of an ABC transporter, the ABCB1, is often associated with MDR in cancer. Previously it was observed that hydantoin compounds can modulate the activity of the ABCB1 pump. Materials and Methods: Fourteen hydantoin derivatives were synthesized and studied for their capacity to increase accumulation of ethidium bromide (EB) by mouse lymphoma cancer cells that were transfected with the human ABCB1 gene and overexpress the human ABCB1 pump. Results: It was observed that the accumulation of EB by the cells in the presence of four of the newly synthesized hydantoins was strongly increased. Similar but milder effects were also observed for the other seven hydantoins; the remaining three had no activity. Conclusion: The 14 hydantoin compounds studied belong to three different structural groups. Structure–activity relationships were studied and important molecular substituents that were possibly responsible for increased the activity of the molecules were identified. This important information may lead to the continuation of our work and to the future synthesis of more active compounds.
“The global burden of cancer continues to increase largely because of the aging and growth of the world population alongside an increasing adoption of cancer-causing behaviors, particularly smoking, in economically developing countries. Based on the GLOBOCAN 2008 estimates, about 12.7 million cancer cases and 7.6 million cancer deaths are estimated to have occurred in 2008; of these, 56% of the cases and 64% of the deaths occurred in the economically developing world.” (1). Despite huge global investment, the absolute cure of any cancer via chemotherapy is always problematic due to the response of the cancer cell, namely, the development of multidrug resistance (MDR) that makes the cancer cell much less susceptible to the initial anticancer drug as well as to many, if not all, the available anticancer drugs of an expanding armamentarium (2, 3). The mechanism by which the MDR phenotype arises involves, among other mechanisms, the overexpression of the transporter of the cell that extrudes the anticancer drug(s) before they reach their therapeutical targets (4, 5). Most of the transporters responsible for the acquisition of resistance in cancer cells belong to the ATP-binding cassette (ABC) family, which includes the ABCB1 transporter (also known as P-glycoprotein (Pgp-1)), one of the most studied transporters of this group.
This efflux pump, responsible for the MDR phenotype of a large number of cancer cells, can be inhibited by a variety of agents from different origins, for example compounds derived from plants (5, 6-8), phenothiazines (9-17) and hydantoins (18). The effect of hydantoins is rather important because unlike other inhibitors of efflux pumps, they take place at concentrations that are non-toxic. The non-toxicity of these compounds is further illustrated by the large variety of medicinal compounds that are derived from hydantoins and which contain the hydantoin structure (18). Therefore, in order to determine the gamut of activity by hydantoins against the efflux pump of MDR cancer cells, additional hydantoin derivatives have been synthesized. The study reported herein demonstrates that these new hydantoins inhibit the ABCB1 efflux pump of the MDR mouse lymphoma cells transfected with the human ABCB1 gene, further supporting that these compounds have a role to play in the therapy of MDR cancer.
Structure of hydantoin derivatives tested (1A-11A, KF4, HY83 and HY84).
Materials and Methods
Cell lines. L5178 mouse T-cell lymphoma cells (ECACC cat. no. 87111908, U.S. FDA, Silver Spring, MD, USA) were transfected with pHa ABCB1/A retrovirus (19, 20), as described elsewhere (3,17,18,19). The ABCB1-expressing cell lines were selected by culturing the infected cells with 60 ng/ml of colchicine (Sigma-Aldrich Chemie GmbH, Steinheim, Germany) to maintain the MDR phenotype. The human ABCB1 gene-transfected L5178 mouse T-cell lymphoma sub-line was cultured in McCoy's 5A medium (Sigma-Aldrich) supplemented with 10% heat-inactivated horse serum (Sigma-Aldrich), L-glutamine (Sigma-Aldrich) and antibiotics (penicillin, streptomycin) (Sigma-Aldrich) at 37°C and in an atmosphere with 5% CO2.
Compounds and materials. Fourteen hydantoin derivatives were synthesized and evaluated for inhibitory effects on the efflux pump system of the MDR mouse lymphoma cell line. The hydantoin compounds and their structures are listed in Table I. Each compound contained two aromatic rings and compounds were divided into three groups (I-III) according to the position of each aromatic fragment. Group I comprises eleven phenylpiperazine-5,5-dimethylhydantoin derivatives (1A–11A) possessing two terminal (un)substituted phenyl rings within substituents at N1- and N3-positions of the hydantoin. Group II includes a racemic (R,S)-ethyl 2-(3-(2-hydroxy-3-(4-(2-hydroxyethyl)piperazin-1-yl)propyl)-2,5-dioxo-4, 4-diphenylimidazolidin-1-yl)acetate (KF4). Two 5-arylidene derivatives (HY83 and HY84), possessing two benzene moieties connected to each other by an ether linker, belong to group III. Synthesis of compounds KF4, HY83 and HY84 was described earlier (21, 22). Synthesis of compounds 1A –11A was performed based on two-phase alkylation described earlier (23-25) and will be described elsewhere. Purity and identity of the 14 hydantoin derivatives were confirmed by the use of spectroscopic methods (1H-NMR, IR), elemental analysis, melting point measurement and thin-layer chromatography). The compounds were dissolved in dimethyl sulfoxide (DMSO). Ethidium bromide (EB) was purchased from Sigma (Madrid, Spain).
EB accumulation assay. The cells were adjusted to a density of 2×106 cells/ml, centrifuged at 2000×g for 2 minutes and re-suspended in phosphate-buffered saline (PBS) containing 0.6% glucose at pH 7.4. The cell suspension was distributed in 90 μl aliquots into 0.2 ml tubes. The tested compounds were individually added at different concentrations in 5 μl volumes of their stock solutions and the samples incubated for 10 minutes at room temperature (≈5°C). After this incubation, 5 μl (1 mg/l final concentration) of EB (20 mg/l stock solution) were added to the samples, the tubes were placed into a Rotor-Gene™ 3000 thermocycler (Corbett Research, Sydney, Australia) and the fluorescence monitored on a real-time basis. Prior to the assay, the instrument was programmed for temperature (37°C), the appropriate excitation and emission wavelengths of EB (530 nm bandpass and 585 nm highpass, respectively), and the time and number of cycles for the recording of the fluorescence. The results were evaluated by Rotor-Gene Analysis Software 6.1 (Build 93) provided by Corbett Research. A complete description of the method has been previously presented in detail (17-19). The essence of the method is that a progressive increase of fluorescence of EB induced by the compound under study provides an estimate of the inhibition of efflux of EB promoted by that agent.
From the real-time data, the activity of the compound, namely the relative final fluorescence index (RFI) of the last time point (minute 60) of the EB accumulation assay, was calculated according to the formula:
 where RFtreated is the relative fluorescence (RF) at the last time point of EB retention curve in the presence of an inhibitor and RFuntreated is the relative fluorescence at the last time point of the EB retention curve of the untreated control. The greater the difference between the RFtreated and RFuntreated control, the greater the degree of EB accumulated and, therefore, the greater the degree of inhibition of the efflux pump system of the cell by the hydantoin compound. Dividing the RFI by the number of micromoles of the hydantoin compound used in the assay provides the specific activity (SA) of inhibition of the efflux pump of MDR mouse lymphoma cells transfected with the human ABCB1 gene.
where RFtreated is the relative fluorescence (RF) at the last time point of EB retention curve in the presence of an inhibitor and RFuntreated is the relative fluorescence at the last time point of the EB retention curve of the untreated control. The greater the difference between the RFtreated and RFuntreated control, the greater the degree of EB accumulated and, therefore, the greater the degree of inhibition of the efflux pump system of the cell by the hydantoin compound. Dividing the RFI by the number of micromoles of the hydantoin compound used in the assay provides the specific activity (SA) of inhibition of the efflux pump of MDR mouse lymphoma cells transfected with the human ABCB1 gene.
Results
The effects of hydantoins on the accumulation of EB by the MDR mouse lymphoma cells transfected with the human ABCB1 gene that codes for the Pgp-1 transporter responsible for the MDR phenotype of the cell is illustrated by Figure 1. The example presented in Figure 1 shows the curve in the presence of HY84. It is readily seen that in the presence of 20 mg/l of HY84, accumulation of EB increases per unit period of time. By the end of the 60-minute assay, the amount of accumulation of EB reflected by the RF reaches the maximum limit of the detection system of the instrument. Each of the 14 hydantoins were similarly evaluated at the same concentration of 20 mg/l for activity against the Pgp-1 transporter and the data converted to SA in RFI/μmoles in order that the effects of each hydantoin on the transporter may be directly compared (Figure 2). As evident from Figure 2, hydantoins HY83, HY84, 2A and 7A had the greatest SA against ABCB1; hydantoins 4A, 6A and 11A had less SA and hydantoins 1A, 3A, 5A and 8A had the lowest SA. Hydantoins KF4, 9A and 10A were inactive against the ABCB1 transporter.
Discussion
The evaluation of each of the new hydantoin derivatives identified those compounds that had activity against the Pgp-1 transporter of mouse lymphoma cells transfected with the human ABCB1 gene. Because the molecular weights of each hydantoin derivative differ, in order to compare the activities against the Pgp-1 transporter, the SA of each compound on a micromolar basis was calculated.
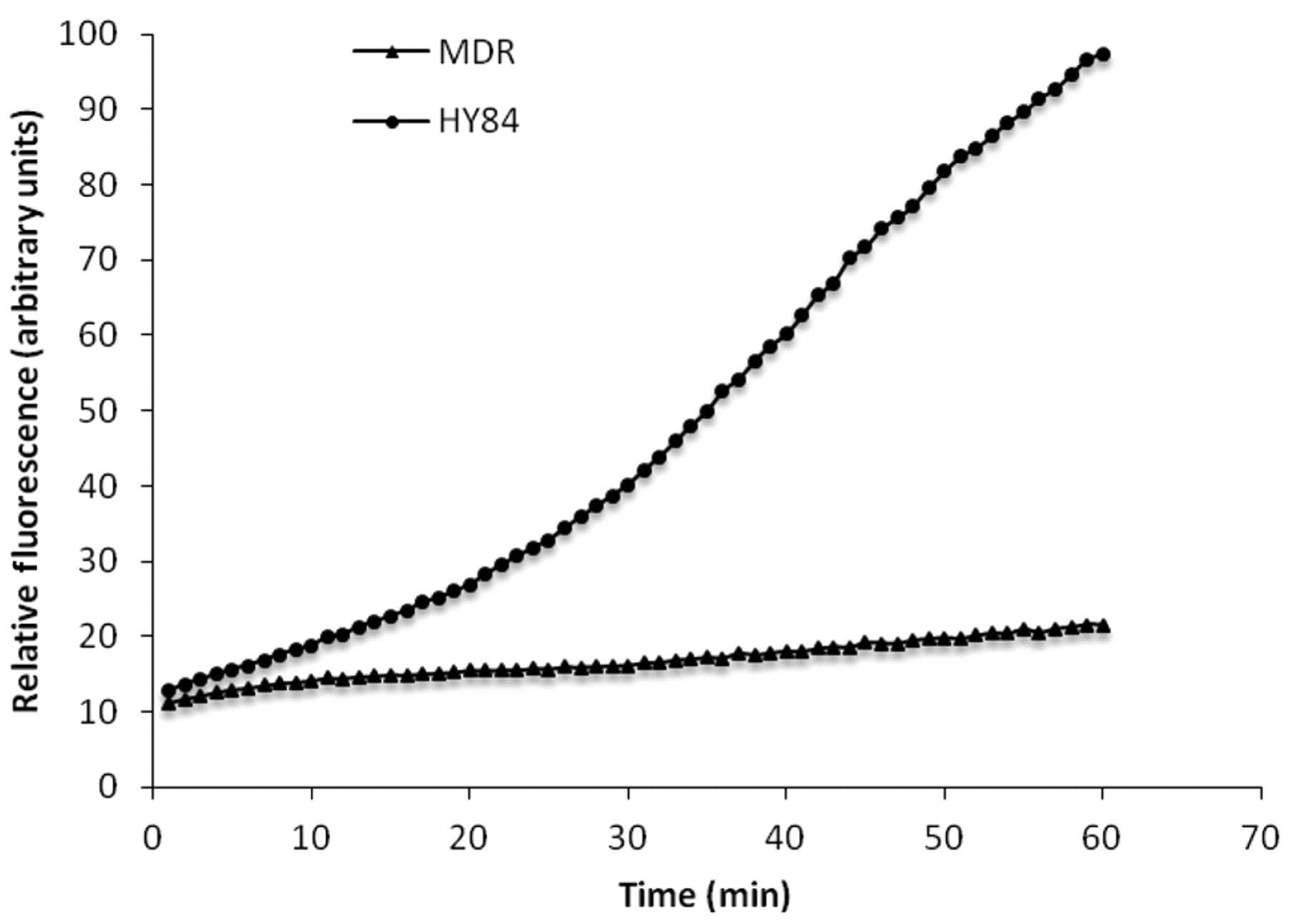
The effect of the hydantoin HY84 on the accumulation of ethidium bromide EB by mouse lymphoma cells transfected with the human ABCB1 gene that codes for the ABCB1 transporter. Mouse lymphoma cells were incubated at 37°C in saline containing 0.6% glucose plus 1 mg/l of EB without and with 20 mg/l of hydantoin HY84.
Structure-activity relationship (SAR) analysis was performed and indicated that a lipophilic benzyloxyarylidene substituent at position 5 of hydantoin (group III) is the most beneficial factor for ABCB1-inhibitory properties of the hydantoin derivatives presented. In the case of phenylpi-perazine derivatives of 3-benzylhydantoin (group I), positions and number of lipophilic substituents at both phenyl rings seem to be crucial for the ABCB1-inhibitory properties. The most favorable is the presence of methoxyl or fluoride substituent at the o-position of the phenylpiperazine phenyl ring (compounds 2A, 4A, 6A and 7A). A comparison of the activities of compounds 1A, 2A and 3A indicated that both the m-position of the methoxyl substituent (3A), as well as an absence of any lipophilic substituent in the aromatic rings (1A), caused a decrease of ABCB1 inhibition by these hydantoin derivatives. Very low ABCB1-inhibitory properties of compounds 9A and 10A also indicated a nonbeneficial impact of m-substitution at phenylpiperazine phenyl ring that can be seen for these structures. Lipophilic substitution (Cl, F) in the p-11A) seems to cause a decrease of the activity but this impact seems to be lower than that of substitution at the m-position. The lowest potency against ABCB1 is observed for compound KF-4 which belongs to group II. Among the 14 hydantoins tested, compound KF-4 is the most hydrophilic because of its hydroxyethylpiperazine terminated fragment, which is probably responsible for the decrease of activity. The SAR analysis performed indicated that benzyloxyarylidene fragments or o-substituted phenylpiperazine phenyl moieties are factors that improve the SA towards ABCB1.

The specific activity (SA) of 14 hydantoin compounds on the Pgp-1 transporter of mouse lymphoma cells transfected with the human ABCB1 gene that codes for the ABCB1 transporter. SA of each hydantoin was calculated by the formula described in the Materials and Methods section.
Hydantoin compounds, such as those previously shown to have activity against the ABCB1 transporter of cancer cells (18), are usually non-toxic. Nevertheless, the hydantoins that had the greatest activity towards the ABCB1 transporter of MDR mouse lymphoma cells transfected with human ABCB1 gene, if they are to be seriously considered as potential candidates for therapy of MDR cancer, must be evaluated for toxicity in an animal model. However, before conducting such in vivo toxicity tests, the hydantoin compounds need to be critically evaluated for their ability to reverse resistance to anticancer agents without themselves affecting the viability of the cell. These studies, as well as a retrospective evaluation of all of the hydantoin compounds studied to date for quantitative SARs, are now in progress and will soon be reported.
Acknowledgements
This study was partly supported by Program K/ZDS/001915 and 501/N-COST/2009/0; COST action BM0701 (K/PMN/000031). A. Martins and G. Spengler acknowledge TÁMOP-4.2.1/B-09/1/KONV-2010-0005 – Creating the Center of Excellence at the University of Szeged supported by the European Union and co-financed by the European Regional Fund. L. Amaral was supported by BCC grant SFRH/BCC/51099/2010 provided by the Fundação para a Ciência e a Tecnologia (FCT) of Portugal. This work was supported by EU-FSE/FEDER-PTDC/BIA-MIC/105509/2008 and EU-FSE/FEDERPTDC/ SAU-FCF/102807/2008 from the Fundação para a Ciência e a Tecnologia of Portugal. This work was partially funded by the Szeged Foundation for Cancer Research.
- Received November 28, 2011.
- Revision received December 19, 2011.
- Accepted December 22, 2011.
- Copyright © 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved




{kind=link}
{kind=link}